Identification of Mouse Cytomegalovirus Resistance Loci by ENU Mutagenesis


Abstract
:1. Introduction
2. A Forward Genetic Approach to Unravel the Host Resistome
3. The innate immune response to MCMV infection
3.1. Molecular mechanisms of MCMV sensing
3.2. Mounting effector functions
3.2.1. The role of pDCs and cDCs in MCMV sensing
3.2.2. Indirect priming of NK cells by DC-secreted cytokines
3.3. NK cells as effectors of the early immune response to MCMV
3.3.2. Elimination of infected cells
3.4. Sensing of MCMV-infected cells by NK cells
3.4.1. The Cmv1 locus
3.4.2. The Cmv2, Cmv3 and Cmv4 loci
4. Screening for resistance loci
4.1. Is Domino susceptibility linked to an alteration of STAT1 configuration?
4.2. Jinx: a step forward in the understanding of a human disease
4.2.1. Is the function of Unc13d cell-specific?

4.2.2. Is FHL-2 distinguishable from FHL-3?
4.3. Warmflash and Moneypennie mutants: new susceptibility phenotypes?
4.4. The MayDay phenotype: unraveling the importance of homeostasis in host survival
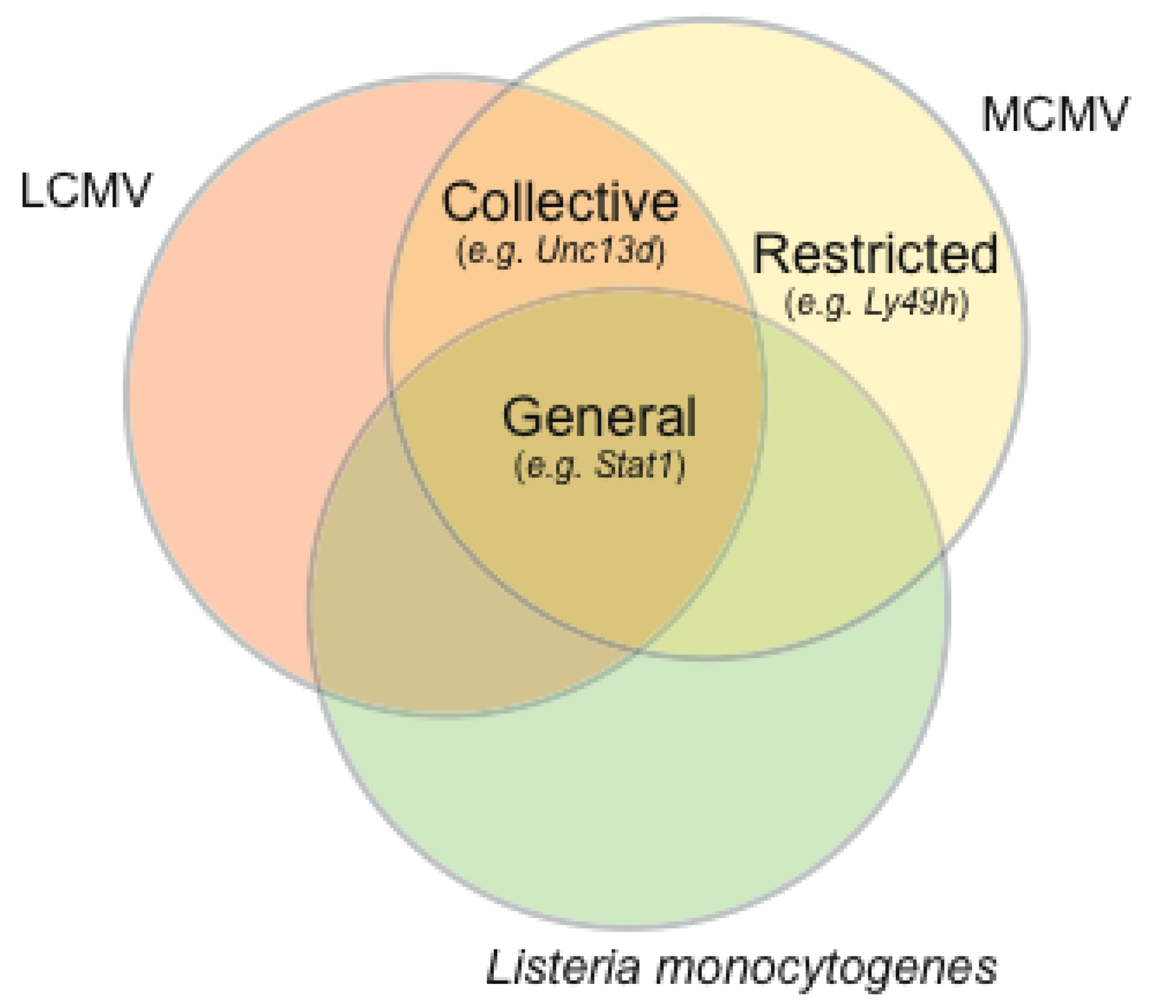
5. The MCMV resistome

{kind=link}
{kind=link}
| Gene name | Protein | Evidence [References] | |
|---|---|---|---|
| Sensors | Ly49h | Ly49H | QTL[56,57] KO[63,64] |
| Tlr3 | TLR3 | KO[9] | |
| Tlr9 | TLR9 | ENU[9] KO[11-13] | |
| Il15rb | IL-15R | Blocking Ab[31] | |
| Ifngr | IFN-gR | KO[109] | |
| Ifnar | IFN-aR | KO[32] | |
| Extracellular signals | Il12b | IL-12 | KO[29,43] |
| Il18 | IL-18 | KO[29,43] | |
| Infg | IFN-g | KO[43] Blocking Ab[43,44] | |
| Mip1a | MIP-1a | KO[110] | |
| Transmitters | Dap12 | KARAP/DAP12 | KO[61,62] |
| Myd88 | MyD88 | KO[9,11,13] | |
| Trif | Trif | ENU[8] | |
| Irf-1 | IRF1 | KO[109] | |
| Stat4 | STAT4 | KO[31] | |
| Stat1 | STAT1 | KO[33,34] ENU[36] | |
| Tyk2 | TYK2 | KO[35] | |
| Effectors | Pfr1 | Perforin | KO[52,101,102] |
| Lyst | Lyst | QTL[111] | |
| Rab27a | Rab27a | QTL (Georgel P., unpublished data) | |
| Unc13d | Unc13d | ENU[82] | |
| Inos | iNos | KO[109] | |
| Homeostasis | Kcnj8 | Kir6.1 | ENU?[103] |

Acknowledgments
References
- Quwailid, M.M.; Hugill, A.; Dear, N.; Vizor, L.; Wells, S.; Horner, E.; Fuller, S.; Weedon, J.; McMath, H.; Woodman, P.; Edwards, D.; Campbell, D.; Rodger, S.; Carey, J.; Roberts, A.; Glenister, P.; Lalanne, Z.; Parkinson, N.; Coghill, E.L.; McKeone, R.; Cox, S.; Willan, J.; Greenfield, A.; Keays, D.; Brady, S.; Spurr, N.; Gray, I.; Hunter, J.; Brown, S.D.; Cox, R.D. A gene-driven ENU-based approach to generating an allelic series in any gene. Mamm. Genome 2004, 15, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, D.; Seburn, K.L.; Wen, G.; Frankel, W.N.; Hamilton, B.A. Mutation rate and predicted phenotypic target sizes in ethylnitrosourea-treated mice. Genetics 2004, 168, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, Y.; Sezutsu, H.; Takahasi, K.R.; Tsuchihashi, K.; Ichikawa, R.; Fujimoto, N.; Kaneko, S.; Nakai, Y.; Uchiyama, M.; Goda, N.; Motoi, R.; Ikeda, A.; Karashima, Y.; Inoue, M.; Kaneda, H.; Masuya, H.; Minowa, O.; Noguchi, H.; Toyoda, A.; Sakaki, Y.; Wakana, S.; Noda, T.; Shiroishi, T.; Gondo, Y. Molecular characterization of ENU mouse mutagenesis and archives. Biochem. Biophys. Res. Commun. 2005, 336, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Hitotsumachi, S.; Carpenter, D.A.; Russell, W.L. Dose-repetition increases the mutagenic effectiveness of N-ethyl-N-nitrosourea in mouse spermatogonia. Proc. Natl. Acad. Sci. U S A 1985, 82, 6619–6621. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.M.; Andoniou, C.E.; Granucci, F.; Ricciardi-Castagnoli, P.; Degli-Esposti, M.A. Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nat. Immunol. 2001, 2, 1077–1084. [Google Scholar] [CrossRef]
- Dalod, M.; Hamilton, T.; Salomon, R.; Salazar-Mather, T.P.; Henry, S.C.; Hamilton, J.D.; Biron, C.A. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J. Exp. Med. 2003, 197, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Pollock, J.L.; Presti, R.M.; Paetzold, S.; Virgin, H.W. Latent murine cytomegalovirus infection in macrophages. Virology 1997, 227, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; Crozat, K.; Sovath, S.; Han, J.; Beutler, B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Tabeta, K.; Georgel, P.; Janssen, E.; Du, X.; Hoebe, K.; Crozat, K.; Mudd, S.; Shamel, L.; Sovath, S.; Goode, J.; Alexopoulou, L.; Flavell, R.A.; Beutler, B. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. U S A 2004, 101, 3516–21. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, K.H.; Richardson-Burns, S.; Alexopoulou, L.; Tyler, K.L.; Flavell, R.A.; Oldstone, M.B. Does Toll-like receptor 3 play a biological role in virus infections? Virology 2004, 322, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Delale, T.; Paquin, A.; Asselin-Paturel, C.; Dalod, M.; Brizard, G.; Bates, E.E.; Kastner, P.; Chan, S.; Akira, S.; Vicari, A.; Biron, C.A.; Trinchieri, G.; Briere, F. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J. Immunol. 2005, 175, 6723–6732. [Google Scholar] [PubMed]
- Krug, A.; French, A.R.; Barchet, W.; Fischer, J.A.; Dzionek, A.; Pingel, J.T.; Orihuela, M.M.; Akira, S.; Yokoyama, W.M.; Colonna, M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004, 21, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Zucchini, N.; Bessou, G.; Traub, S.; Robbins, S.H.; Uematsu, S.; Akira, S.; Alexopoulou, L.; Dalod, M. Cutting edge: Overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J. Immunol. 2008, 180, 5799–5803. [Google Scholar] [PubMed]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; Georgel, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J.; Shamel, L.; Herskovits, A.A.; Portnoy, D.A.; Cooke, M.; Tarantino, L.M.; Wiltshire, T.; Steinberg, B.E.; Grinstein, S.; Beutler, B. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Zhang, S.Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; Lorenzo, L.; Plancoulaine, S.; Senechal, B.; Geissmann, F.; Tabeta, K.; Hoebe, K.; Du, X.; Miller, R.L.; Heron, B.; Mignot, C.; de Villemeur, T.B.; Lebon, P.; Dulac, O.; Rozenberg, F.; Beutler, B.; Tardieu, M.; Abel, L.; Casanova, J.L. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Spooner, E.; Hoebe, K.; Beutler, B.; Ploegh, H.L.; Kim, Y.M. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J. Cell Biol. 2007, 177, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Brinkmann, M.M.; Paquet, M.E.; Ploegh, H.L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 2008, 452, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Fukui, R.; Saitoh, S.I.; Matsumoto, F.; Kozuka-Hata, H.; Oyama, M.; Tabeta, K.; Beutler, B.; Miyake, K. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J. Exp. Med. 2009, 206, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Ahmad-Nejad, P.; Hacker, H.; Rutz, M.; Bauer, S.; Vabulas, R.M.; Wagner, H. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 2002, 32, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, T.; DeFranco, A.L. Ligand-regulated chimeric receptor approach reveals distinctive subcellular localization and signaling properties of the Toll-like receptors. J. Biol. Chem. 2004, 279, 19008–19017. [Google Scholar] [CrossRef] [PubMed]
- Allman, D.; Dalod, M.; Asselin-Paturel, C.; Delale, T.; Robbins, S.H.; Trinchieri, G.; Biron, C.A.; Kastner, P.; Chan, S. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood 2006, 108, 4025–4034. [Google Scholar] [CrossRef] [PubMed]
- Andoniou, C.E.; van Dommelen, S.L.; Voigt, V.; Andrews, D.M.; Brizard, G.; Asselin-Paturel, C.; Delale, T.; Stacey, K.J.; Trinchieri, G.; Degli-Esposti, M.A. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat. Immunol. 2005, 6, 1011–1019. [Google Scholar] [CrossRef]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Dokun, A.O.; Chu, D.T.; Yang, L.; Bendelac, A.S.; Yokoyama, W.M. Analysis of in situ NK cell responses during viral infection. J. Immunol. 2001, 167, 5286–5293. [Google Scholar] [PubMed]
- Andrews, D.M.; Scalzo, A.A.; Yokoyama, W.M.; Smyth, M.J.; Degli-Esposti, M.A. Functional interactions between dendritic cells and NK cells during viral infection. Nat. Immunol. 2003, 4, 175–181. [Google Scholar] [CrossRef]
- Robbins, S.H.; Bessou, G.; Cornillon, A.; Zucchini, N.; Rupp, B.; Ruzsics, Z.; Sacher, T.; Tomasello, E.; Vivier, E.; Koszinowski, U.H.; Dalod, M. Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog. 2007, 3, e123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orange, J.S.; Wang, B.; Terhorst, C.; Biron, C.A. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J. Exp. Med. 1995, 182, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S.; Biron, C.A. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J. Immunol. 1996, 156, 1138–1142. [Google Scholar] [PubMed]
- Pien, G.C.; Satoskar, A.R.; Takeda, K.; Akira, S.; Biron, C.A. Cutting edge: selective IL-18 requirements for induction of compartmental IFN-gamma responses during viral infection. J. Immunol. 2000, 165, 4787–4791. [Google Scholar] [PubMed]
- Chaix, J.; Tessmer, M.S.; Hoebe, K.; Fuseri, N.; Ryffel, B.; Dalod, M.; Alexopoulou, L.; Beutler, B.; Brossay, L.; Vivier, E.; Walzer, T. Cutting edge: Priming of NK cells by IL-18. J. Immunol. 2008, 181, 1627–1631. [Google Scholar] [PubMed]
- Nguyen, K.B.; Salazar-Mather, T.P.; Dalod, M.Y.; Van Deusen, J.B.; Wei, X.Q.; Liew, F.Y.; Caligiuri, M.A.; Durbin, J.E.; Biron, C.A. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 2002, 169, 4279–4287. [Google Scholar] [PubMed]
- Salazar-Mather, T.P.; Lewis, C.A.; Biron, C.A. Type I interferons regulate inflammatory cell trafficking and macrophage inflammatory protein 1alpha delivery to the liver. J. Clin. Invest. 2002, 110, 321–330. [Google Scholar] [PubMed]
- Durbin, J.E.; Hackenmiller, R.; Simon, M.C.; Levy, D.E. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 1996, 84, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Rao, D.T.; Gertner, R.; Gimeno, R.; Frey, A.B.; Levy, D.E. Distinct requirements for IFNs and STAT1 in NK cell function. J. Immunol. 2000, 165, 3571–3577. [Google Scholar] [PubMed]
- Strobl, B.; Bubic, I.; Bruns, U.; Steinborn, R.; Lajko, R.; Kolbe, T.; Karaghiosoff, M.; Kalinke, U.; Jonjic, S.; Muller, M. Novel functions of tyrosine kinase 2 in the antiviral defense against murine cytomegalovirus. J. Immunol. 2005, 175, 4000–4008. [Google Scholar] [PubMed]
- Crozat, K.; Georgel, P.; Rutschmann, S.; Mann, N.; Du, X.; Hoebe, K.; Beutler, B. Analysis of the MCMV resistome by ENU mutagenesis. Mamm. Genome 2006, 17, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S.; Biron, C.A. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J. Immunol. 1996, 156, 4746–4756. [Google Scholar] [PubMed]
- Lodolce, J.P.; Boone, D.L.; Chai, S.; Swain, R.E.; Dassopoulos, T.; Trettin, S.; Ma, A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity 1998, 9, 669–676. [Google Scholar] [CrossRef]
- Kennedy, M.K.; Glaccum, M.; Brown, S.N.; Butz, E.A.; Viney, J.L.; Embers, M.; Matsuki, N.; Charrier, K.; Sedger, L.; Willis, C.R.; Brasel, K.; Morrissey, P.J.; Stocking, K.; Schuh, J.C.; Joyce, S.; Peschon, J.J. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med. 2000, 191, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, J.F.; Warner, J.F.; Dennert, G.; Welsh, R.M. Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo. J. Exp. Med. 1985, 161, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Byron, K.S.; Sullivan, J.L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989, 320, 1731–1735. [Google Scholar] [PubMed]
- Welsh, R.M.; Brubaker, J.O.; Vargas-Cortes, M.; O'Donnell, C.L. Natural killer (NK) cell response to virus infections in mice with severe combined immunodeficiency. The stimulation of NK cells and the NK cell-dependent control of virus infections occur independently of T and B cell function. J. Exp. Med. 1991, 173, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Mather, T.P.; Hokeness, K.L. Calling in the troops: regulation of inflammatory cell trafficking through innate cytokine/chemokine networks. Viral Immuno.l 2003, 16, 291–306. [Google Scholar] [CrossRef]
- Presti, R.M.; Pollock, J.L.; Dal Canto, A.J.; O'Guin, A.K.; Virgin IV, H.W. Interferon gamma regulates acute and latent murine cytomegalovirus infection and chronic disease of the great vessels. J. Exp. Med. 1998, 188, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Heise, M.T.; Virgin, H.W. The T-cell-independent role of gamma interferon and tumor necrosis factor alpha in macrophage activation during murine cytomegalovirus and herpes simplex virus infections. J. Virol. 1995, 69, 904–909. [Google Scholar] [PubMed]
- Hengel, H.; Lucin, P.; Jonjic, S.; Ruppert, T.; Koszinowski, U.H. Restoration of cytomegalovirus antigen presentation by gamma interferon combats viral escape. J. Virol. 1994, 68, 289–297. [Google Scholar] [PubMed]
- Gribaudo, G.; Ravaglia, S.; Caliendo, A.; Cavallo, R.; Gariglio, M.; Martinotti, M.G.; Landolfo, S. Interferons inhibit onset of murine cytomegalovirus immediate-early gene transcription. Virology 1993, 197, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Lucin, P.; Jonjic, S.; Messerle, M.; Polic, B.; Hengel, H.; Koszinowski, U.H. Late phase inhibition of murine cytomegalovirus replication by synergistic action of interferon-gamma and tumour necrosis factor. J. Gen. Virol. 1994, 75, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, E.K.; Wu, X.; Hammer, J.A.; Henkart, P.A. Defective granule exocytosis in Rab27a-deficient lymphocytes from Ashen mice. J. Cell Biol. 2001, 152, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Pao, L.I.; Sumaria, N.; Kelly, J.M.; van Dommelen, S.; Cretney, E.; Wallace, M.E.; Anthony, D.A.; Uldrich, A.P.; Godfrey, D.I.; Papadimitriou, J.M.; Mullbacher, A.; Degli-Esposti, M.A.; Smyth, M.J. Functional analysis of granzyme M and its role in immunity to infection. J. Immunol. 2005, 175, 3235–3243. [Google Scholar] [PubMed]
- van Dommelen, S.L.; Sumaria, N.; Schreiber, R.D.; Scalzo, A.A.; Smyth, M.J.; Degli-Esposti, M.A. Perforin and granzymes have distinct roles in defensive immunity and immunopathology. Immunity 2006, 25, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.; Chu, D.T.; O'Guin, A.K.; Yokoyama, W.M.; Virgin, H.W. Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J. Virol. 2005, 79, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Dokun, A.O.; Kim, S.; Smith, H.R.; Kang, H.S.; Chu, D.T.; Yokoyama, W.M. Specific and nonspecific NK cell activation during virus infection. Nat. Immunol. 2001, 2, 951–956. [Google Scholar] [CrossRef]
- French, A.R.; Sjolin, H.; Kim, S.; Koka, R.; Yang, L.; Young, D.A.; Cerboni, C.; Tomasello, E.; Ma, A.; Vivier, E.; Karre, K.; Yokoyama, W.M. DAP12 signaling directly augments proproliferative cytokine stimulation of NK cells during viral infections. J. Immunol. 2006, 177, 4981–4990. [Google Scholar] [PubMed]
- Scalzo, A.A.; Fitzgerald, N.A.; Simmons, A.; La Vista, A.B.; Shellam, G.R. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J. Exp. Med. 1990, 171, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Girard, S.; Macina, D.; Busa, M.; Zafer, A.; Belouchi, A.; Gros, P.; Vidal, S.M. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat. Genet. 2001, 28, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.G.; Dokun, A.O.; Heusel, J.W.; Smith, H.R.; Beckman, D.L.; Blattenberger, E.A.; Dubbelde, C.E.; Stone, L.R.; Scalzo, A.A.; Yokoyama, W.M. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 2001, 292, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Daniels, K.A.; Devora, G.; Lai, W.C.; O'Donnell, C.L.; Bennett, M.; Welsh, R.M. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J. Exp. Med. 2001, 194, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Arase, H.; Mocarski, E.S.; Campbell, A.E.; Hill, A.B.; Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 2002, 296, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.R.; Heusel, J.W.; Mehta, I.K.; Kim, S.; Dorner, B.G.; Naidenko, O.V.; Iizuka, K.; Furukawa, H.; Beckman, D.L.; Pingel, J.T.; Scalzo, A.A.; Fremont, D.H.; Yokoyama, W.M. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. U S A 2002, 99, 8826–8831. [Google Scholar] [PubMed]
- Sjolin, H.; Tomasello, E.; Mousavi-Jazi, M.; Bartolazzi, A.; Karre, K.; Vivier, E.; Cerboni, C. Pivotal role of KARAP/DAP12 adaptor molecule in the natural killer cell-mediated resistance to murine cytomegalovirus infection. J. Exp. Med. 2002, 195, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Orr, M. T.; Sun, J.C.; Hesslein, D.G.; Arase, H.; Phillips, J.H.; Takai, T.; Lanier, L.L. Ly49H signaling through DAP10 is essential for optimal natural killer cell responses to mouse cytomegalovirus infection. J. Exp. Med. 2009, 206, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.P.; French, A.R.; Plougastel, B.F.; Pingel, J.T.; Orihuela, M.M.; Buller, M.L.; Yokoyama, W.M. Ly49h is necessary for genetic resistance to murine cytomegalovirus. Immunogenetics 2008, 60, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Fodil-Cornu, N.; Lee, S.H.; Belanger, S.; Makrigiannis, A.P.; Biron, C.A.; Buller, R.M.; Vidal, S.M. Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J. Immunol. 2008, 181, 6394–6405. [Google Scholar] [PubMed]
- Scalzo, A.A.; Manzur, M.; Forbes, C.A.; Brown, M.G.; Shellam, G.R. NK gene complex haplotype variability and host resistance alleles to murine cytomegalovirus in wild mouse populations. Immunol. Cell Biol. 2005, 83, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Booth, T.W.; Scalzo, A.A.; Carrello, C.; Lyons, P.A.; Farrell, H.E.; Singleton, G.R.; Shellam, G.R. Molecular and biological characterization of new strains of murine cytomegalovirus isolated from wild mice. Arch Virol. 1993, 132, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Voigt, V.; Forbes, C.A.; Tonkin, J.N.; Degli-Esposti, M.A.; Smith, H.R.; Yokoyama, W.M.; Scalzo, A.A. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc. Natl. Acad. Sci. U S A 2003, 100, 13483–13488. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.H.; Guseva, N.V.; Ball, B.L.; Heusel, J.W. Characterization of murine cytomegalovirus m157 from infected cells and identification of critical residues mediating recognition by the NK cell receptor Ly49H. J. Immunol. 2008, 181, 265–275. [Google Scholar] [PubMed]
- Brown, M.G.; Scalzo, A.A.; Stone, L.R.; Clark, P.Y.; Du, Y.; Palanca, B.; Yokoyama, W.M. Natural killer gene complex (Nkc) allelic variability in inbred mice: evidence for Nkc haplotypes. Immunogenetics 2001, 53, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Sabastian, P.; Clark, P.; Brown, M.G. Cmv1-independent antiviral role of NK cells revealed in murine cytomegalovirus-infected New Zealand White mice. J. Immunol. 2004, 173, 6312–6318. [Google Scholar] [PubMed]
- Chalmer, J.E.; Mackenzie, J.S.; Stanley, N.F. Resistance to murine cytomegalovirus linked to the major histocompatibility complex of the mouse. J. Gen. Virol. 1977, 37, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Dighe, A.; Rodriguez, M.; Sabastian, P.; Xie, X.; McVoy, M.; Brown, M.G. Requisite H2k role in NK cell-mediated resistance in acute murine cytomegalovirus-infected MA/My mice. J. Immunol. 2005, 175, 6820–6828. [Google Scholar] [PubMed]
- Desrosiers, M.P.; Kielczewska, A.; Loredo-Osti, J.C.; Adam, S.G.; Makrigiannis, A.P.; Lemieux, S.; Pham, T.; Lodoen, M.B.; Morgan, K.; Lanier, L.L.; Vidal, S.M. Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat. Genet. 2005, 37, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Kielczewska, A.; Pyzik, M.; Sun, T.; Krmpotic, A.; Lodoen, M.B.; Munks, M.W.; Babic, M.; Hill, A.B.; Koszinowski, U.H.; Jonjic, S.; Lanier, L.L.; Vidal, S.M. Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J. Exp. Med. 2009, 206, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Dighe, A.; Clark, P.; Sabastian, P.; Buss, S.; Brown, M.G. Deficient major histocompatibility complex-linked innate murine cytomegalovirus immunity in MA/My.L-H2b mice and viral downregulation of H-2k class I proteins. J. Virol. 2007, 81, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.G.; Caraux, A.; Fodil-Cornu, N.; Loredo-Osti, J.C.; Lesjean-Pottier, S.; Jaubert, J.; Bubic, I.; Jonjic, S.; Guenet, J.L.; Vidal, S.M.; Colucci, F. Cmv4, a new locus linked to the NK cell gene complex, controls innate resistance to cytomegalovirus in wild-derived mice. J. Immunol. 2006, 176, 5478–5485. [Google Scholar] [PubMed]
- Belardelli, F.; Gessani, S.; Proietti, E.; Locardi, C.; Borghi, P.; Watanabe, Y.; Kawade, Y.; Gresser, I. Studies on the expression of spontaneous and induced interferons in mouse peritoneal macrophages by means of monoclonal antibodies to mouse interferons. J. Gen. Virol. 1987, 68, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Haan, S.; Kortylewski, M.; Behrmann, I.; Muller-Esterl, W.; Heinrich, P.C.; Schaper, F. Cytoplasmic STAT proteins associate prior to activation. Biochem. J. 2000, 345, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ren, Z.; Parker, G.N.; Sondermann, H.; Pastorello, M.A.; Wang, W.; McMurray, J.S.;Demeler; Darnell, J.E.; Chen, X. Structural bases of unphosphorylated STAT1 association and receptor binding . Mol. Cell 2005, 17, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Henriksen, M. A.; Takeuchi, K.; Schaefer, O.; Liu, B.; ten Hoeve, J.; Ren, Z.; Mao, X.; Chen, X.; Shuai, K.; Darnell, J. E. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc. Natl. Acad. Sci. U S A 2005, 102, 3966–3971. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Crozat, K.; Hoebe, K.; Ugolini, S.; Hong, N. A.; Janssen, E.; Rutschmann, S.; Mudd, S.; Sovath, S.; Vivier, E.; Beutler, B. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp Med 2007, 204, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, J.; Callebaut, I.; Raposo, G.; Certain, S.; Bacq, D.; Dumont, C.; Lambert, N.; Ouachee-Chardin, M.; Chedeville, G.; Tamary, H.; Minard-Colin, V.; Vilmer, E.; Blanche, S.; Le Deist, F.; Fischer, A.; de Saint Basile, G. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003, 115, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Marcenaro, S.; Gallo, F.; Martini, S.; Santoro, A.; Griffiths, G.M.; Arico, M.; Moretta, L.; Pende, D. Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13-4 defect and discriminates between genetic subtypes of the disease. Blood 2006, 108, 2316–2323. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.; Bossi, G.; Griffiths, G.M. Linking albinism and immunity: the secrets of secretory lysosomes. Science 2004, 305, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Whitmire, J.K.; Tan, J.T.; Whitton, J.L. Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J. Exp. Med. 2005, 201, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.M.; Burke, B.A.; McKenna, R.W.; McClain, K.L.; White, J.G.; Nesbit, M.E.; Filipovich, A.H. The accelerated phase of Chediak-Higashi syndrome. An expression of the virus-associated hemophagocytic syndrome? Cancer 1985, 56, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Menasche, G.; Pastural, E.; Feldmann, J.; Certain, S.; Ersoy, F.; Dupuis, S.; Wulffraat, N.; Bianchi, D.; Fischer, A.; Le Deist, F.; de Saint Basile, G. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet. 2000, 25, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Pachlopnik Schmid, J.; Ho, C.H.; Diana, J.; Pivert, G.; Lehuen, A.; Geissmann, F.; Fischer, A.; de Saint Basile, G. A Griscelli syndrome type 2 murine model of hemophagocytic lymphohistiocytosis (HLH). Eur. J. Immunol. 2008, 38, 3219–3225. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.M. Serotonin deficiency and prolonged bleeding in beige mice. Proc. Soc. Exp. Biol. Med. 1976, 151, 32–39. [Google Scholar] [PubMed]
- Novak, E.K.; Hui, S.W.; Swank, R.T. Platelet storage pool deficiency in mouse pigment mutations associated with seven distinct genetic loci. Blood 1984, 63, 536–544. [Google Scholar] [PubMed]
- Novak, E.K.; Reddington, M.; Zhen, L.; Stenberg, P.E.; Jackson, C.W.; McGarry, M.P.; Swank, R.T. Inherited thrombocytopenia caused by reduced platelet production in mice with the gunmetal pigment gene mutation. Blood 1995, 85, 1781–1789. [Google Scholar] [PubMed]
- Zhen, L.; Jiang, S.; Feng, L.; Bright, N.A.; Peden, A.A.; Seymour, A.B.; Novak, E.K.; Elliott, R.; Gorin, M.B.; Robinson, M.S.; Swank, R.T. Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood 1999, 94, 146–155. [Google Scholar] [PubMed]
- Wilson, S.M.; Yip, R.; Swing, D.A.; O'Sullivan, T.N.; Zhang, Y.; Novak, E.K.; Swank, R.T.; Russell, L.B.; Copeland, N.G.; Jenkins, N.A. A mutation in Rab27a causes the vesicle transport defects observed in ashen mice. Proc. Natl. Acad. Sci. U S A 2000, 97, 7933–7938. [Google Scholar] [CrossRef] [PubMed]
- Brzezinska, A.A.; Johnson, J.L.; Munafo, D.B.; Crozat, K.; Beutler, B.; Kiosses, W.B.; Ellis, B.A.; Catz, S.D. The Rab27a effectors JFC1/Slp1 and Munc13-4 regulate exocytosis of neutrophil granules. Traffic 2008, 9, 2151–2164. [Google Scholar] [CrossRef]
- Pivot-Pajot, C.; Varoqueaux, F.; de Saint Basile, G.; Bourgoin, S.G. Munc13-4 regulates granule secretion in human neutrophils. J. Immunol. 2008, 180, 6786–6797. [Google Scholar] [PubMed]
- Stepp, S.E.; Dufourcq-Lagelouse, R.; Le Deist, F.; Bhawan, S.; Certain, S.; Mathew, P.A.; Henter, J.I.; Bennett, M.; Fischer, A.; de Saint Basile, G.; Kumar, V. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999, 286, 1957–1959. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.B.; Hildeman, D.; Kappler, J.; Marrack, P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 2004, 104, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Griffiths, G.M. Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nat. Med. 1999, 5, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Blott, E.J.; Griffiths, G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002, 3, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.H.; Welsh, R.M. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J. Virol. 1997, 71, 267–275. [Google Scholar] [PubMed]
- van Dommelen, S.L.; Tabarias, H.A.; Smyth, M.J.; Degli-Esposti, M.A. Activation of natural killer (NK) T cells during murine cytomegalovirus infection enhances the antiviral response mediated by NK cells. J. Virol. 2003, 77, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.; Crozat, K.; Berger, M.; Xia, Y.; Sovath, S.; Schaffer, L.; Eleftherianos, I.; Imler, J.L.; Beutler, B. ATP-sensitive potassium channels mediate survival during infection in mammals and insects. Nat. Genet. 2007, 39, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Ruzek, M.C.; Miller, A.H.; Opal, S.M.; Pearce, B.D.; Biron, C.A. Characterization of early cytokine responses and an interleukin (IL)-6-dependent pathway of endogenous glucocorticoid induction during murine cytomegalovirus infection. J. Exp. Med. 1997, 185, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, A.; Rosner, E.; Lanning, J.; Parachuru, L.; Dhar Chowdhury, P.; Han, S.; Lopez, G.; Tong, X.; Yoshida, H.; Nakamura, T.Y.; Artman, M.; Giblin, J.P.; Tinker, A.; Coetzee, W.A. Immunolocalization of KATP channel subunits in mouse and rat cardiac myocytes and the coronary vasculature. BMC Physiol. 2005, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutler, B.; Crozat, K.; Koziol, J.A.; Georgel, P. Genetic dissection of innate immunity to infection: the mouse cytomegalovirus model. Curr. Opin. Immunol. 2005, 17, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Georgel, P.; Rutschmann, S.; Jiang, Z.; Croker, B.; Crozat, K. Genetic analysis of innate resistance to mouse cytomegalovirus (MCMV). Brief Funct. Genomic Proteomic 2005, 4, 203–213. [Google Scholar] [CrossRef]
- Wang, F.Z.; Weber, F.; Croce, C.; Liu, C.G.; Liao, X.; Pellett, P.E. Human cytomegalovirus infection alters the expression of cellular microRNA species that affect its replication. J. Virol. 2008, 82, 9065–9074. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.A.; Rodrigues, E.G.; Tsuji, M. Multifactorial protective mechanisms to limit viral replication in the lung of mice during primary murine cytomegalovirus infection. Viral Immunol. 2000, 13, 287–295. [Google Scholar] [PubMed]
- Salazar-Mather, T.P.; Orange, J.S.; Biron, C.A. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1alpha (MIP-1alpha)-dependent pathways. J. Exp. Med. 1998, 187, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shellam, G.R.; Allan, J.E.; Papadimitriou, J.M.; Bancroft, G.J. Increased susceptibility to cytomegalovirus infection in beige mutant mice. Proc. Natl. Acad. Sci. U S A 1981, 78, 5104–5108. [Google Scholar] [CrossRef] [PubMed]
- Ruzek, M.C.; Pearce, B.D.; Miller, A.H.; Biron, C.A. Endogenous glucocorticoids protect against cytokine-mediated lethality during viral infection. J. Immunol. 1999, 162, 3527–3533. [Google Scholar] [PubMed]
- Kucic, N.; Mahmutefendic, H.; Lucin, P. Inhibition of protein kinases C prevents murine cytomegalovirus replication. J. Gen. Virol. 2005, 86, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Fodil-Cornu, N.; Kozij, N.; Wu, Q.; Rozen, R.; Vidal, S.M. Methylenetetrahydrofolate reductase (MTHFR) deficiency enhances resistance against cytomegalovirus infection. Genes Immun. 2009, (in press). [Google Scholar]
- Schleiss, M.R. Cytomegalovirus vaccines: at last, a major step forward. Herpes 2009, 15, 44–45. [Google Scholar] [PubMed]
- Griffiths, P.D.; Walter, S. Cytomegalovirus. Curr. Opin. Infect. Dis. 2005, 18, 241–245. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Crozat, K.; Georgel, P. Identification of Mouse Cytomegalovirus Resistance Loci by ENU Mutagenesis. Viruses 2009, 1, 460-483. https://doi.org/10.3390/v1030460
Crozat K, Georgel P. Identification of Mouse Cytomegalovirus Resistance Loci by ENU Mutagenesis. Viruses. 2009; 1(3):460-483. https://doi.org/10.3390/v1030460
Chicago/Turabian StyleCrozat, Karine, and Philippe Georgel. 2009. "Identification of Mouse Cytomegalovirus Resistance Loci by ENU Mutagenesis" Viruses 1, no. 3: 460-483. https://doi.org/10.3390/v1030460