Transcriptomics Reveal Antiviral Gene Induction in the Egyptian Rousette Bat Is Antagonized In Vitro by Marburg Virus Infection

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Virus Infection
2.3. RNA Isolation
2.4. Design of the ERB NanoString nCounter CodeSet
2.5. nCounter Hybridization and Data Collection
2.6. nCounter Analysis
2.7. RNA-Seq Data Collection
2.8. RNA-Seq Analysis
2.9. Pathway Analysis
2.10. Data Visualization
2.11. GTEx Data Analysis
3. Results
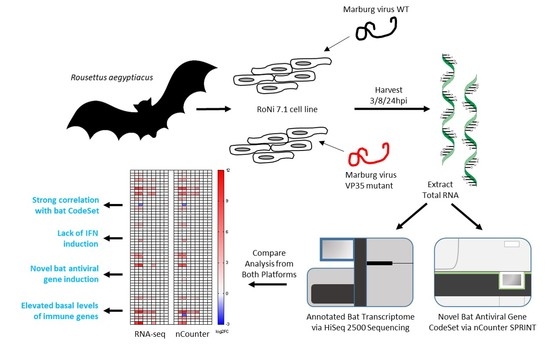
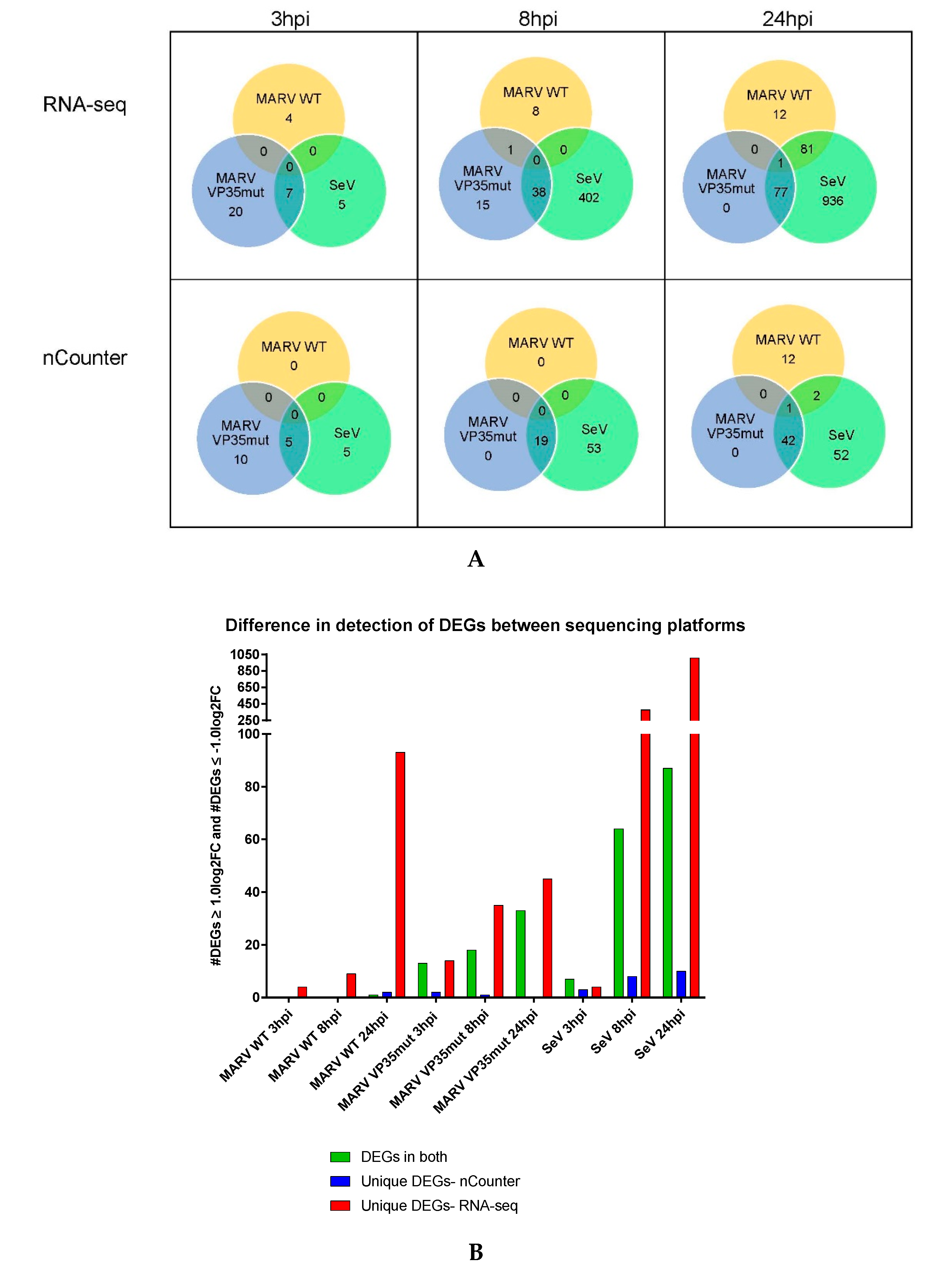
3.1. Differential Gene Expression for the nCounter CodeSet Strongly Correlates to RNA-Seq
3.2. MARV WT Does Not Induce a Canonical Antiviral Response Gene Program in RoNi Cells
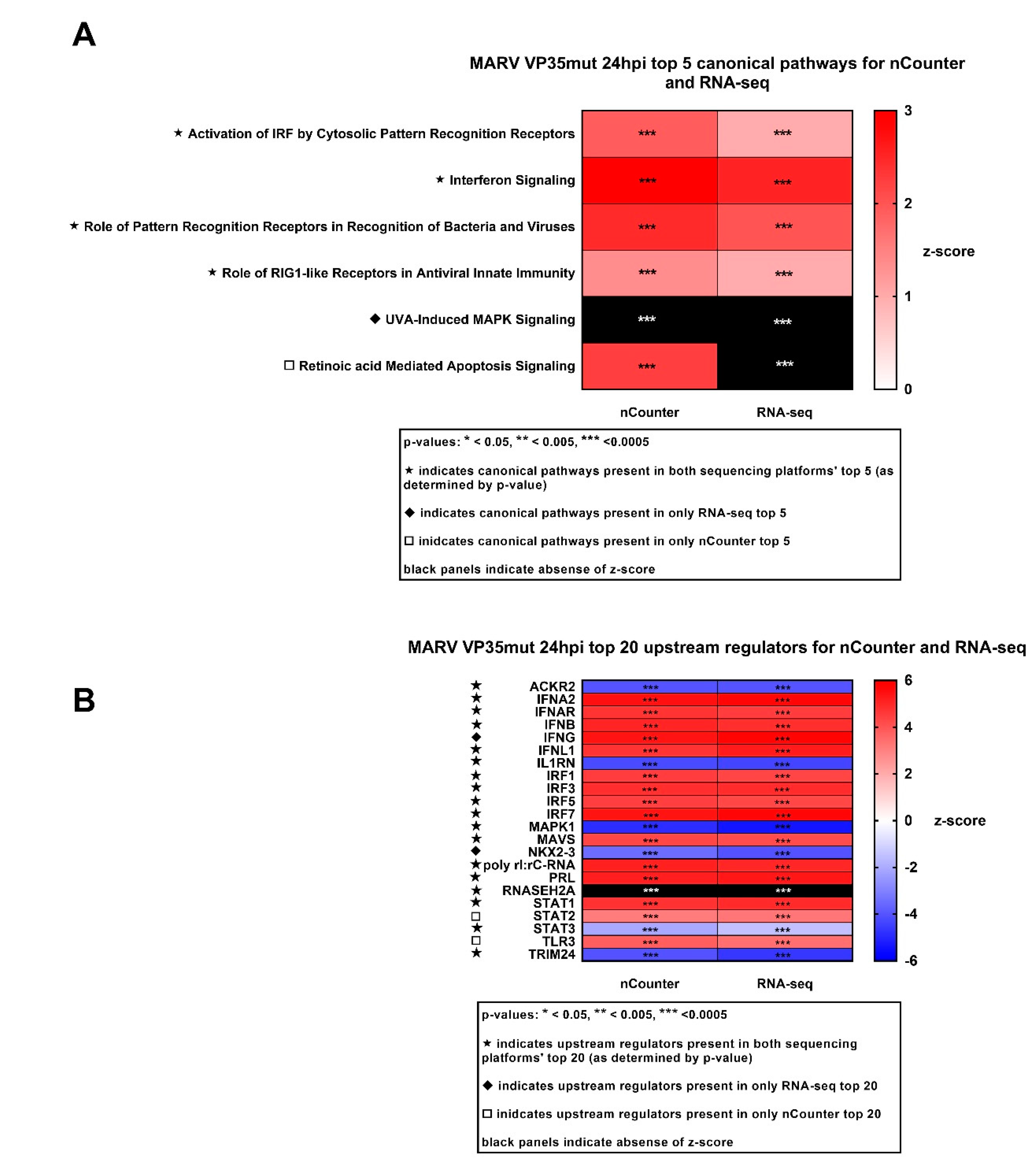
3.3. IFN Antagonism-Deficient MARV Infection Shows Robust Upregulation of Innate Immune Response Genes
3.4. Expanded Immune Gene Repertoire Is Upregulated in RoNi Cells
3.5. Basal Expression of Many Antiviral Genes Is Higher in Uninfected RoNi Cells Compared to Human Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Halpin, K.; Hyatt, A.D.; Fogarty, R.; Middleton, D.; Bingham, J.; Epstein, J.H.; Rahman, S.A.; Hughes, T.; Smith, C.; Field, H.E.; et al. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: A comprehensive experimental study of virus transmission. Am. J. Trop. Med. Hyg. 2011, 85, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Amman, B.R.; Swanepoel, R.; Nichol, S.T.; Towner, J.S. Ecology of Filoviruses. Curr. Top. Microbiol. Immunol. 2017, 411, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Kortepeter, M.G.; Bausch, D.G.; Bray, M. Basic clinical and laboratory features of filoviral hemorrhagic fever. J. Infect. Dis. 2011, 204 (Suppl. 3), S810–S816. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Pourrut, X.; Albarino, C.G.; Nkogue, C.N.; Bird, B.H.; Grard, G.; Ksiazek, T.G.; Gonzalez, J.P.; Nichol, S.T.; Leroy, E.M. Marburg virus infection detected in a common African bat. PLoS ONE 2007, 2, e764. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Carroll, S.A.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef] [PubMed]
- Swanepoel, R.; Smit, S.B.; Rollin, P.E.; Formenty, P.; Leman, P.A.; Kemp, A.; Burt, F.J.; Grobbelaar, A.A.; Croft, J.; Bausch, D.G.; et al. Studies of reservoir hosts for Marburg virus. Emerg. Infect. Dis. 2007, 13, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Schuh, A.J.; Amman, B.R.; Jones, M.E.; Sealy, T.K.; Uebelhoer, L.S.; Spengler, J.R.; Martin, B.E.; Coleman-McCray, J.A.; Nichol, S.T.; Towner, J.S. Modelling filovirus maintenance in nature by experimental transmission of Marburg virus between Egyptian rousette bats. Nat. Commun. 2017, 8, 14446. [Google Scholar] [CrossRef] [PubMed]
- Amman, B.R.; Jones, M.E.; Sealy, T.K.; Uebelhoer, L.S.; Schuh, A.J.; Bird, B.H.; Coleman-McCray, J.D.; Martin, B.E.; Nichol, S.T.; Towner, J.S. Oral shedding of Marburg virus in experimentally infected Egyptian fruit bats (Rousettus aegyptiacus). J. Wildl. Dis. 2015, 51, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.E.; Schuh, A.J.; Amman, B.R.; Sealy, T.K.; Zaki, S.R.; Nichol, S.T.; Towner, J.S. Experimental Inoculation of Egyptian Rousette Bats (Rousettus aegyptiacus) with Viruses of the Ebolavirus and Marburgvirus Genera. Viruses 2015, 7, 3420–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paweska, J.T.; Jansen van Vuren, P.; Masumu, J.; Leman, P.A.; Grobbelaar, A.A.; Birkhead, M.; Clift, S.; Swanepoel, R.; Kemp, A. Virological and serological findings in Rousettus aegyptiacus experimentally inoculated with vero cells-adapted hogan strain of Marburg virus. PLoS ONE 2012, 7, e45479. [Google Scholar] [CrossRef] [PubMed]
- Lubaki, N.M.; Younan, P.; Santos, R.I.; Meyer, M.; Iampietro, M.; Koup, R.A.; Bukreyev, A. The Ebola Interferon Inhibiting Domains Attenuate and Dysregulate Cell-Mediated Immune Responses. PLoS Pathog. 2016, 12, e1006031. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Mühlberger, E.; Carter, V.; Grosch, M.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Weber, F.; Klenk, H.-D.; Katze, M.G. Global Suppression of the Host Antiviral Response by Ebola- and Marburgviruses: Increased Antagonism of the Type I Interferon Response Is Associated with Enhanced Virulence. J. Virol. 2006, 80, 3009–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guito, J.C.; Albarino, C.G.; Chakrabarti, A.K.; Towner, J.S. Novel activities by ebolavirus and marburgvirus interferon antagonists revealed using a standardized in vitro reporter system. Virology 2017, 501, 147–165. [Google Scholar] [CrossRef] [PubMed]
- Ilinykh, P.A.; Lubaki, N.M.; Widen, S.G.; Renn, L.A.; Theisen, T.C.; Rabin, R.L.; Wood, T.G.; Bukreyev, A. Different Temporal Effects of Ebola Virus VP35 and VP24 Proteins on Global Gene Expression in Human Dendritic Cells. J. Virol. 2015, 89, 7567–7583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, B.C.; Basler, C.F. Effects of Filovirus Interferon Antagonists on Responses of Human Monocyte-Derived Dendritic Cells to RNA Virus Infection. J. Virol. 2016, 90, 5108–5118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmin, I.V.; Schwarz, T.M.; Ilinykh, P.A.; Jordan, I.; Ksiazek, T.G.; Sachidanandam, R.; Basler, C.F.; Bukreyev, A. Innate Immune Responses of Bat and Human Cells to Filoviruses: Commonalities and Distinctions. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Krahling, V.; Amman, F.; Barth, E.; Bernhart, S.H.; Carmelo, V.A.; Collatz, M.; Doose, G.; Eggenhofer, F.; Ewald, J.; et al. Differential transcriptional responses to Ebola and Marburg virus infection in bat and human cells. Sci. Rep. 2016, 6, 34589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, A.L.; Bird, B.H.; Towner, J.S.; Antoniadou, Z.A.; Zaki, S.R.; Nichol, S.T. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J. Virol. 2008, 82, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Tachedjian, M.; Wynne, J.W.; Boyd, V.; Cui, J.; Smith, I.; Cowled, C.; Ng, J.H.; Mok, L.; Michalski, W.P.; et al. Contraction of the type I IFN locus and unusual constitutive expression of IFN-alpha in bats. Proc. Natl. Acad. Sci. USA 2016, 113, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, A.; Hoffmann, M.; Muller, M.A.; Munster, V.J.; Gnirss, K.; Kiene, M.; Tsegaye, T.S.; Behrens, G.; Herrler, G.; Feldmann, H.; et al. Comparative analysis of Ebola virus glycoprotein interactions with human and bat cells. J. Infect. Dis. 2011, 204 (Suppl. 3), S840–S849. [Google Scholar] [CrossRef] [PubMed]
- Pavlovich, S.S.; Lovett, S.P.; Koroleva, G.; Guito, J.C.; Arnold, C.E.; Nagle, E.R.; Kulcsar, K.; Lee, A.; Thibaud-Nissen, F.; Hume, A.J.; et al. The Egyptian Rousette Genome Reveals Unexpected Features of Bat Antiviral Immunity. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.K.; Kulcsar, K.A.; Elliott, O.; Khiabanian, H.; Nagle, E.R.; Jones, M.E.; Amman, B.R.; Sanchez-Lockhart, M.; Towner, J.S.; Palacios, G.; et al. De novo transcriptome reconstruction and annotation of the Egyptian rousette bat. BMC Genom. 2015, 16, 1033. [Google Scholar] [CrossRef] [PubMed]
- Albarino, C.G.; Uebelhoer, L.S.; Vincent, J.P.; Khristova, M.L.; Chakrabarti, A.K.; McElroy, A.; Nichol, S.T.; Towner, J.S. Development of a reverse genetics system to generate recombinant Marburg virus derived from a bat isolate. Virology 2013, 446, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albarino, C.G.; Wiggleton Guerrero, L.; Spengler, J.R.; Uebelhoer, L.S.; Chakrabarti, A.K.; Nichol, S.T.; Towner, J.S. Recombinant Marburg viruses containing mutations in the IID region of VP35 prevent inhibition of Host immune responses. Virology 2015, 476, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Boil. 2002, 3, Research0034. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Feagins, A.R.; Basler, C.F. Lloviu virus VP24 and VP35 proteins function as innate immune antagonists in human and bat cells. Virology 2015, 485, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Mikulasova, A.; Martinez-Sobrido, L.; Paragas, J.; Muhlberger, E.; Bray, M.; Klenk, H.D.; Palese, P.; Garcia-Sastre, A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003, 77, 7945–7956. [Google Scholar] [CrossRef] [PubMed]
- Schumann, M.; Gantke, T.; Muhlberger, E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J. Virol. 2009, 83, 8993–8997. [Google Scholar] [CrossRef] [PubMed]
- Bale, S.; Julien, J.P.; Bornholdt, Z.A.; Krois, A.S.; Wilson, I.A.; Saphire, E.O. Ebolavirus VP35 coats the backbone of double-stranded RNA for interferon antagonism. J. Virol. 2013, 87, 10385–10388. [Google Scholar] [CrossRef] [PubMed]
- Luthra, P.; Ramanan, P.; Mire, C.E.; Weisend, C.; Tsuda, Y.; Yen, B.; Liu, G.; Leung, D.W.; Geisbert, T.W.; Ebihara, H.; et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 2013, 14, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.W.; Park, M.S.; Martinez, O.; Valmas, C.; Lopez, C.B.; Basler, C.F. Ebolavirus VP35 suppresses IFN production from conventional but not plasmacytoid dendritic cells. Immunol. Cell Boil. 2011, 89, 792–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.L.; Schountz, T.; Wang, L.F. Antiviral immune responses of bats: A review. Zoonoses Public Health 2013, 60, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Yaddanapudi, K.; Palacios, G.; Towner, J.S.; Chen, I.; Sariol, C.A.; Nichol, S.T.; Lipkin, W.I. Implication of a retrovirus-like glycoprotein peptide in the immunopathogenesis of Ebola and Marburg viruses. FASEB J. 2006, 20, 2519–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, S.; Huye, L.E.; Pagano, J.S. Regulation of the transcriptional activity of the IRF7 promoter by a pathway independent of interferon signaling. J. Boil. Chem. 2005, 280, 12262–12270. [Google Scholar] [CrossRef] [PubMed]
- Sariol, C.A.; Munoz-Jordan, J.L.; Abel, K.; Rosado, L.C.; Pantoja, P.; Giavedoni, L.; Rodriguez, I.V.; White, L.J.; Martinez, M.; Arana, T.; et al. Transcriptional activation of interferon-stimulated genes but not of cytokine genes after primary infection of rhesus macaques with dengue virus type 1. Clin. Vaccine Immunol. 2007, 14, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Servant, M.J.; Grandvaux, N.; Hiscott, J. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem. Pharmacol. 2002, 64, 985–992. [Google Scholar] [CrossRef]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Pitha, P.M.; Harty, R.N. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3974–3979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malakhova, O.A.; Zhang, D.E. ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J. Boil. Chem. 2008, 283, 8783–8787. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz-Rivera, P.C.; Kanchwala, M.; Liang, H.; Kumar, A.; Wang, L.F.; Xing, C.; Schoggins, J.W. The IFN Response in Bats Displays Distinctive IFN-Stimulated Gene Expression Kinetics with Atypical RNASEL Induction. J. Immunol. 2018, 200, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, D.-E. Interferon-Stimulated Gene 15 and the Protein ISGylation System. J. Interferon Cytokine Res. 2011, 31, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroya-Beltri, C.; Guerra, S.; Sánchez-Madrid, F. ISGylation—A key to lock the cell gates for preventing the spread of threats. J. Cell Sci. 2017, 130, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Samarajiwa, S.A.; Mangan, N.E.; Hardy, M.P.; Najdovska, M.; Dubach, D.; Braniff, S.-J.; Owczarek, C.M.; Hertzog, P.J. Soluble IFN Receptor Potentiates In Vivo Type I IFN Signaling and Exacerbates TLR4-Mediated Septic Shock. J. Immunol. 2014, 192, 4425–4435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Yang, S.W.; Park, J.M.; Ka, S.H.; Kim, J.-H.; Kong, Y.-Y.; Jeon, Y.J.; Seol, J.H.; Chung, C.H. Positive feedback regulation of p53 transactivity by DNA damage-induced ISG15 modification. Nat. Commun. 2016, 7, 12513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummer, B.T.; Li, X.L.; Hassel, B.A. Role for p53 in gene induction by double-stranded RNA. J. Virol. 2001, 75, 7774–7777. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnold, C.E.; Guito, J.C.; Altamura, L.A.; Lovett, S.P.; Nagle, E.R.; Palacios, G.F.; Sanchez-Lockhart, M.; Towner, J.S. Transcriptomics Reveal Antiviral Gene Induction in the Egyptian Rousette Bat Is Antagonized In Vitro by Marburg Virus Infection. Viruses 2018, 10, 607. https://doi.org/10.3390/v10110607
Arnold CE, Guito JC, Altamura LA, Lovett SP, Nagle ER, Palacios GF, Sanchez-Lockhart M, Towner JS. Transcriptomics Reveal Antiviral Gene Induction in the Egyptian Rousette Bat Is Antagonized In Vitro by Marburg Virus Infection. Viruses. 2018; 10(11):607. https://doi.org/10.3390/v10110607
Chicago/Turabian StyleArnold, Catherine E., Jonathan C. Guito, Louis A. Altamura, Sean P. Lovett, Elyse R. Nagle, Gustavo F. Palacios, Mariano Sanchez-Lockhart, and Jonathan S. Towner. 2018. "Transcriptomics Reveal Antiviral Gene Induction in the Egyptian Rousette Bat Is Antagonized In Vitro by Marburg Virus Infection" Viruses 10, no. 11: 607. https://doi.org/10.3390/v10110607