Mammarenaviral Infection Is Dependent on Directional Exposure to and Release from Polarized Intestinal Epithelia


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses and Titration Assay
2.2. Polarization of Caco-2 Cells and Infection of Polarized Cells
2.3. Confocal Microscopy
2.4. Attachment Assay
2.5. Statistical Analyses
3. Results
3.1. Infection of Polarized Caco-2 Cells with OW Arenaviruses Does Not Affect the Monolayer Integrity
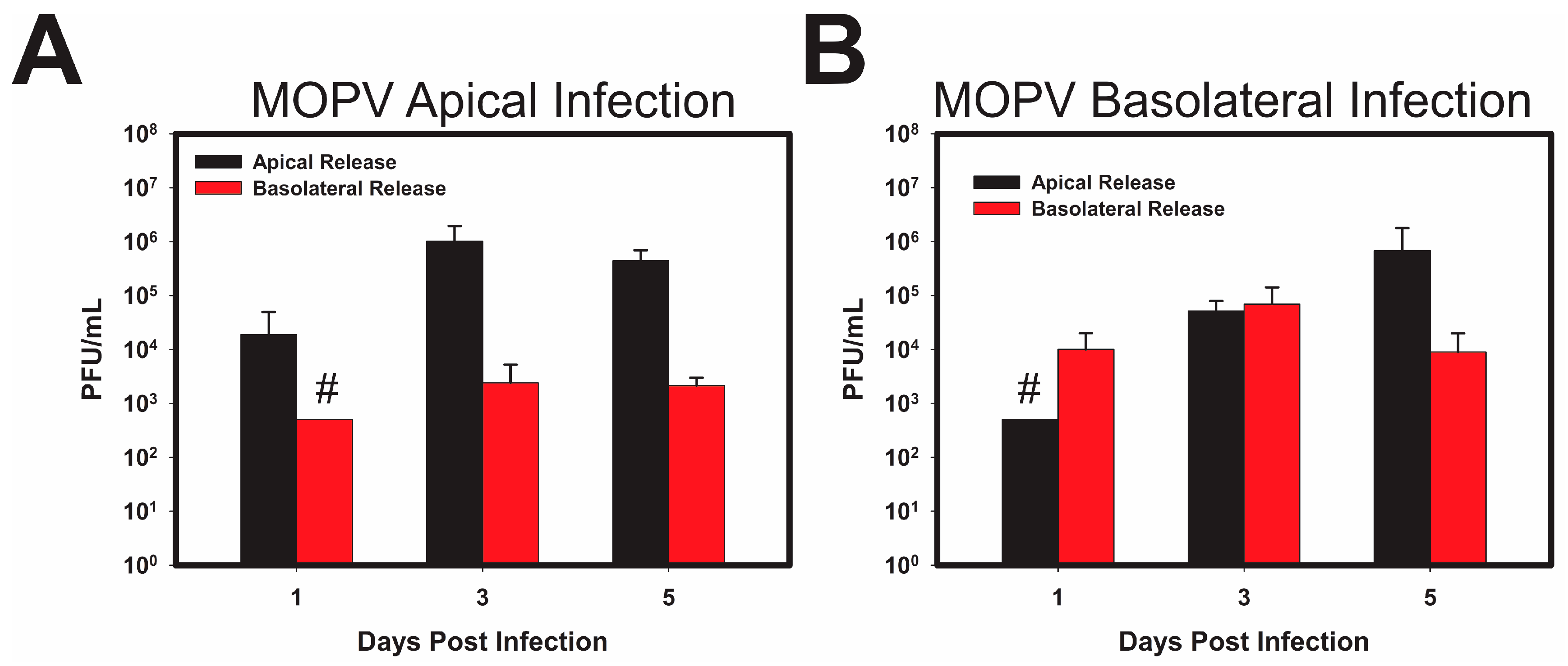
3.2. LASV/MOPV Reassortant ML-29 Exhibits Different Viral Entry and Exit Patterns Compared to Either LCMV or MOPV
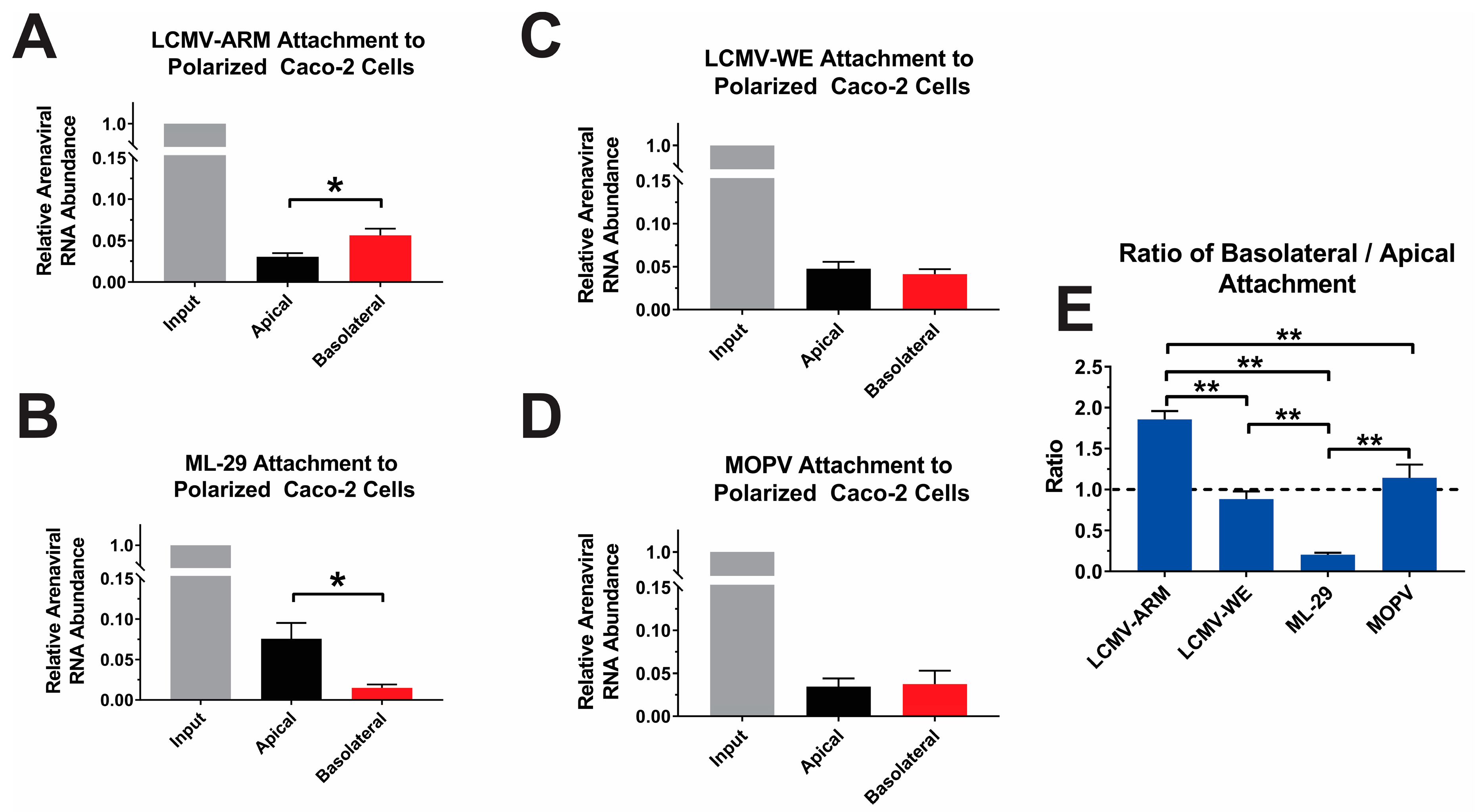
3.3. Attachment and Binding of OW Arenaviruses to Polarized Caco-2 Cells
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murphy, B.R.; Webster, R. Fields Virology; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1996. [Google Scholar]
- Bodewes, R.; Kik, M.J.; Raj, V.S.; Schapendonk, C.M.; Haagmans, B.L.; Smits, S.L.; Osterhaus, A.D. Detection of novel divergent arenaviruses in boid snakes with inclusion body disease in The Netherlands. J. Gen. Virol. 2013, 94 Pt 6, 1206–1210. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Bào, Y.; Buchmeier, M.J.; Charrel, R.N.; Clawson, A.N.; Clegg, C.S.; DeRisi, J.L.; Emonet, S.; Gonzalez, J.P.; Kuhn, J.H.; et al. Past, present, and future of arenavirus taxonomy. Arch. Virol. 2015, 160, 1851–1874. [Google Scholar] [CrossRef] [PubMed]
- Bowen, M.D.; Peters, C.J.; Nichol, S.T. The phylogeny of New World (Tacaribe complex) arenaviruses. Virology 1996, 219, 285–290. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa fever. Curr. Top. Microbiol. Immunol. 2002, 262, 75–109. [Google Scholar] [PubMed]
- Khan, S.H.; Goba, A.; Chu, M.; Roth, C.; Healing, T.; Marx, A.; Fair, J.; Guttieri, M.C.; Ferro, P.; Imes, T.; et al. New opportunities for field research on the pathogenesis and treatment of Lassa fever. Antivir. Res. 2008, 78, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Fichet-Calvet, E.; Rogers, D.J. Risk maps of Lassa fever in West Africa. PLoS Negl. Trop. Dis. 2009, 3, e388. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, M.E.; Wilda, M.; Levingston Macleod, J.M.; D’Antuono, A.; Foscaldi, S.; Marino Buslje, C.; Lopez, N. Molecular determinants of arenavirus Z protein homo-oligomerization and L polymerase binding. J. Virol. 2011, 85, 12304–12314. [Google Scholar] [CrossRef] [PubMed]
- Richmond, J.K.; Baglole, D.J. Lassa fever: Epidemiology, clinical features, and social consequences. BMJ Br. Med. J. (Clin. Res. Ed.) 2003, 327, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Shapiro, B.J.; Matranga, C.B.; Sealfon, R.; Lin, A.E.; Moses, L.M.; Folarin, O.A.; Goba, A.; Odia, I.; Ehiane, P.E.; et al. Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus. Cell 2015, 162, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Ter Meulen, J.; Lukashevich, I.; Sidibe, K.; Inapogui, A.; Marx, M.; Dorlemann, A.; Yansane, M.L.; Koulemou, K.; Chang-Claude, J.; Schmitz, H. Hunting of peridomestic rodents and consumption of their meat as possible risk factors for rodent-to-human transmission of Lassa virus in the Republic of Guinea. Am. J. Trop. Med. Hyg. 1996, 55, 661–666. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Scribner, C.L.; Craven, R.B.; Johnson, K.M.; Elliott, L.H.; Belmont-Williams, R. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986, 314, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Goicochea, M.A.; Zapata, J.C.; Bryant, J.; Davis, H.; Salvato, M.S.; Lukashevich, I.S. Evaluation of Lassa virus vaccine immunogenicity in a CBA/J-ML29 mouse model. Vaccine 2012, 30, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S.; Patterson, J.; Carrion, R.; Moshkoff, D.; Ticer, A.; Zapata, J.; Brasky, K.; Geiger, R.; Hubbard, G.B.; Bryant, J.; et al. A live attenuated vaccine for Lassa fever made by reassortment of Lassa and Mopeia viruses. J. Virol. 2005, 79, 13934–13942. [Google Scholar] [CrossRef] [PubMed]
- Barkar, N.D.; Lukashevich, I.S. Lassa and Mozambique viruses: Cross protection in experiments on mice and action of immunosuppressants on experimental infections. Vopr. Virusol. 1989, 34, 598–603. [Google Scholar] [PubMed]
- Carrion, R., Jr.; Patterson, J.L.; Johnson, C.; Gonzales, M.; Moreira, C.R.; Ticer, A.; Brasky, K.; Hubbard, G.B.; Moshkoff, D.; Zapata, J.; et al. A ML29 reassortant virus protects guinea pigs against a distantly related Nigerian strain of Lassa virus and can provide sterilizing immunity. Vaccine 2007, 25, 4093–4102. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S.; Carrion, R., Jr.; Salvato, M.S.; Mansfield, K.; Brasky, K.; Zapata, J.; Cairo, C.; Goicochea, M.; Hoosien, G.E.; Ticer, A.; et al. Safety, immunogenicity, and efficacy of the ML29 reassortant vaccine for Lassa fever in small non-human primates. Vaccine 2008, 26, 5246–5254. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.C.; Poonia, B.; Bryant, J.; Davis, H.; Ateh, E.; George, L.; Crasta, O.; Zhang, Y.; Slezak, T.; Jaing, C.; et al. An attenuated Lassa vaccine in SIV-infected rhesus macaques does not persist or cause arenavirus disease but does elicit Lassa virus-specific immunity. Virol. J. 2013, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Carrion, R., Jr.; Brasky, K.; Mansfield, K.; Johnson, C.; Gonzales, M.; Ticer, A.; Lukashevich, I.; Tardif, S.; Patterson, J. Lassa virus infection in experimentally infected marmosets: Liver pathology and immunophenotypic alterations in target tissues. J. Virol. 2007, 81, 6482–6490. [Google Scholar] [CrossRef] [PubMed]
- Moshkoff, D.A.; Salvato, M.S.; Lukashevich, I.S. Molecular characterization of a reassortant virus derived from Lassa and Mopeia viruses. Virus Genes 2007, 34, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Montali, R.J.; Scanga, C.A.; Pernikoff, D.; Wessner, D.R.; Ward, R.; Holmes, K.V. A common-source outbreak of callitrichid hepatitis in captive tamarins and marmosets. J. Infect. Dis. 1993, 167, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Montali, R.J.; Connolly, B.M.; Armstrong, D.L.; Scanga, C.A.; Holmes, K.V. Pathology and immunohistochemistry of callitrichid hepatitis, an emerging disease of captive New World primates caused by lymphocytic choriomeningitis virus. Am. J. Pathol. 1995, 147, 1441–1449. [Google Scholar] [PubMed]
- Lukashevich, I.S.; Djavani, M.; Rodas, J.D.; Zapata, J.C.; Usborne, A.; Emerson, C.; Mitchen, J.; Jahrling, P.B.; Salvato, M.S. Hemorrhagic Fever occurs after intravenous but not after intragastric inoculation of rhesus macaques with LCMV. J. Med. Virol. 2002, 67, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Rodas, J.D.; Lukashevich, I.S.; Zapata, J.C.; Cairo, C.; Tikhonov, I.; Djavani, M.; Pauza, C.D.; Salvato, M.S. Mucosal arenavirus infection of primates can protect them from lethal hemorrhagic fever. J. Med. Virol. 2004, 72, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, E.H.; Larson, E.W.; Dominik, J.W. Effect of environmental factors on aerosol-induced Lassa virus infection. J. Med. Virol. 1984, 14, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Schlie, K.; Maisa, A.; Freiberg, F.; Groseth, A.; Strecker, T.; Garten, W. Viral protein determinants of Lassa virus entry and release from polarized epithelial cells. J. Virol. 2010, 84, 3178–3188. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.T.; Tseng, J.; Perrone, L.; Worthy, M.; Popov, V.; Peters, C.J. Apical entry and release of severe acute respiratory syndrome-associated coronavirus in polarized Calu-3 lung epithelial cells. J. Virol. 2005, 79, 9470–9479. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, E.; Zeier, M. Hantavirus causing hemorrhagic fever with renal syndrome enters from the apical surface and requires decay-accelerating factor (DAF/CD55). J. Virol. 2008, 82, 4257–4264. [Google Scholar] [CrossRef] [PubMed]
- Excoffon, K.J.; Guglielmi, K.M.; Wetzel, J.D.; Gansemer, N.D.; Campbell, J.A.; Dermody, T.S.; Zabner, J. Reovirus preferentially infects the basolateral surface and is released from the apical surface of polarized human respiratory epithelial cells. J. Infect. Dis. 2008, 197, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Dylla, D.E.; Michele, D.E.; Campbell, K.P.; McCray, P.B., Jr. Basolateral entry and release of New and Old World arenaviruses from human airway epithelia. J. Virol. 2008, 82, 6034–6038. [Google Scholar] [CrossRef] [PubMed]
- Esclatine, A.; Lemullois, M.; Servin, A.L.; Quero, A.M.; Geniteau-Legendre, M. Human cytomegalovirus infects Caco-2 intestinal epithelial cells basolaterally regardless of the differentiation state. J. Virol. 2000, 74, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Compans, R.W. Polarized entry of canine parvovirus in an epithelial cell line. J. Virol. 1989, 63, 3164–3167. [Google Scholar] [PubMed]
- Lukashevich, I.S.; Vasiuchkov, A.D.; Stel’makh, T.A.; Scheslenok, E.P.; Shabanov, A.G. The isolation and characteristics of reassortants between the Lassa and Mopeia arenaviruses. Vopr. Virusol. 1991, 36, 146–150. [Google Scholar] [PubMed]
- Lukashevich, I.S. Generation of reassortants between African arenaviruses. Virology 1992, 188, 600–605. [Google Scholar] [CrossRef]
- Hayes, M.W.; Carrion, R., Jr.; Nunneley, J.; Medvedev, A.E.; Salvato, M.S.; Lukashevich, I.S. Pathogenic Old World arenaviruses inhibit TLR2/Mal-dependent proinflammatory cytokines in vitro. J. Virol. 2012, 86, 7216–7226. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Mori, A.; Satsu, H.; Shimizu, M. New model for studying the migration of immune cells into intestinal epithelial cell monolayers. Cytotechnology 2003, 43, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Grasset, E.; Pinto, M.; Dussaulx, E.; Zweibaum, A.; Desjeux, J.F. Epithelial properties of human colonic carcinoma cell line Caco-2: Electrical parameters. Am. J. Physiol. Cell Physiol. 1984, 247 Pt 1, C260–C267. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.K.; Cheung, D.S.; Wu, M.S.; Warner, T.F.; Salvato, M.S. Murine infection with lymphocytic choriomeningitis virus following gastric inoculation. J. Virol. 1996, 70, 7213–7218. [Google Scholar] [PubMed]
- Ter Meulen, J.; Badusche, M.; Kuhnt, K.; Doetze, A.; Satoguina, J.; Marti, T.; Loeliger, C.; Koulemou, K.; Koivogui, L.; Schmitz, H.; et al. Characterization of human CD4(+) T-cell clones recognizing conserved and variable epitopes of the Lassa virus nucleoprotein. J. Virol. 2000, 74, 2186–2192. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.B.; Walker, D.H.; King, I.J.; Webb, P.A.; Elliott, L.H.; Whitfield, S.G.; Johnson, K.M. Lassa virus hepatitis: A study of fatal Lassa fever in humans. Am. J. Trop. Med. Hyg. 1986, 35, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Oppliger, J.; Torriani, G.; Herrador, A.; Kunz, S. Lassa Virus Cell Entry via Dystroglycan Involves an Unusual Pathway of Macropinocytosis. J. Virol. 2016, 90, 6412–6429. [Google Scholar] [CrossRef] [PubMed]
- Torriani, G.; Galan-Navarro, C.; Kunz, S. Lassa Virus Cell Entry Reveals New Aspects of Virus-Host Cell Interaction. J. Virol. 2017, 91, e01902-16. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Ngo, N.; de la Torre, J.C. Sodium hydrogen exchangers contribute to arenavirus cell entry. J. Virol. 2014, 88, 643–654. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warner, N.L.; Jokinen, J.D.; Beier, J.I.; Sokoloski, K.J.; Lukashevich, I.S. Mammarenaviral Infection Is Dependent on Directional Exposure to and Release from Polarized Intestinal Epithelia. Viruses 2018, 10, 75. https://doi.org/10.3390/v10020075
Warner NL, Jokinen JD, Beier JI, Sokoloski KJ, Lukashevich IS. Mammarenaviral Infection Is Dependent on Directional Exposure to and Release from Polarized Intestinal Epithelia. Viruses. 2018; 10(2):75. https://doi.org/10.3390/v10020075
Chicago/Turabian StyleWarner, Nikole L., Jenny D. Jokinen, Juliane I. Beier, Kevin J. Sokoloski, and Igor S. Lukashevich. 2018. "Mammarenaviral Infection Is Dependent on Directional Exposure to and Release from Polarized Intestinal Epithelia" Viruses 10, no. 2: 75. https://doi.org/10.3390/v10020075
APA StyleWarner, N. L., Jokinen, J. D., Beier, J. I., Sokoloski, K. J., & Lukashevich, I. S. (2018). Mammarenaviral Infection Is Dependent on Directional Exposure to and Release from Polarized Intestinal Epithelia. Viruses, 10(2), 75. https://doi.org/10.3390/v10020075