The Intersection of HPV Epidemiology, Genomics and Mechanistic Studies of HPV-Mediated Carcinogenesis


 ,
,
Abstract
:
1. Background
2. Recent Discoveries in HPV Genomics
3. Summary/Next Steps: Molecular Mechanisms Underlying HPV Carcinogenesis
3.1. Viral Molecules and Their Interactions with Host Cellular Machinery
3.2. Tissue Tropism and Site of Infection
3.3. Regulation of HPV Transcription
3.4. HPV Integration into Host Genomes
3.5. Viral–Host Interactions
4. Synthesizing Current Knowledge and Moving Forward in the Era of NGS, Systems Biology, and Big Data
4.1. Defining HPV “Fitness”
4.2. HPV Genome Annotation and Other New Emerging Data Concepts
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global Burden of Human Papillomavirus and Related Diseases. Vaccine 2012, 30 (Suppl. 5), F12–F23. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Cancer Collaboration. The global burden of cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar]
- Schiffman, M.; Herrero, R.; Desalle, R.; Hildesheim, A.; Wacholder, S.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Morales, J.; Guillen, D.; et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology 2005, 337, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Bernard, H.U.; Ong, C.K.; Chan, S.P.; Hofmann, B.; Delius, H. Phylogenetic analysis of 48 papillomavirus types and 28 subtypes and variants: A showcase for the molecular evolution of DNA viruses. J. Virol. 1992, 66, 5714–5725. [Google Scholar] [PubMed]
- Yamada, T.; Manos, M.M.; Peto, J.; Greer, C.E.; Munoz, N.; Bosch, F.X.; Wheeler, C.M. Human papillomavirus type 16 sequence variation in cervical cancers: A worldwide perspective. J. Virol. 1997, 71, 2463–2472. [Google Scholar] [PubMed]
- Pimenoff, V.N.; de Oliveira, C.M.; Bravo, I.G. Transmission between Archaic and Modern Human Ancestors during the Evolution of the Oncogenic Human Papillomavirus 16. Mol. Biol. Evol. 2017, 34, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; van Doorslaer, K.; DeSalle, R.; Wood, C.E.; Kaplan, J.R.; Wagner, J.D.; Burk, R.D. Genomic diversity and interspecies host infection of alpha12 Macaca fascicularis papillomaviruses (MfPVs). Virology 2009, 393, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K. Evolution of the papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U. Regulatory elements in the viral genome. Virology 2013, 445, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Burk, R.D.; Harari, A.; Chen, Z. Human papillomavirus genome variants. Virology 2013, 445, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Howell-Jones, R.; Li, N.; Bruni, L.; de Sanjosé, S.; Franceschi, S.; Clifford, G.M. Human papillomavirus types in 115,789 HPV-positive women: A meta-analysis from cervical infection to cancer. Int. J. Cancer 2012, 131, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- De Sanjose, S.; Quint, W.G.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.R.; et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- Serrano, B.; de Sanjosé, S.; Tous, S.; Quiros, B.; Muñoz, N.; Bosch, X.; Alemany, L. Human papillomavirus genotype attribution for HPVs 6, 11, 16, 18, 31, 33, 45, 52 and 58 in female anogenital lesions. Eur. J. Cancer 2015, 51, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Bunge, E.; Bakker, M.; Castellsagué, X. The incidence, clearance and persistence of non-cervical human papillomavirus infections: A systematic review of the literature. BMC Infect. Dis. 2016, 16, 293. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsagué, X.; Laporte, L.; Bosch, F.X.; de Sanjosé, S.; Trottier, H. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, Z.; Wentzensen, N.; Zhang, X.; Yu, K.; Yang, Q.; Mitchell, J.; et al. HPV16 Sublineage Associations with Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J. Natl. Cancer Inst. 2016, 108, djw100. [Google Scholar] [CrossRef] [PubMed]
- Cornet, I.; Gheit, T.; Iannacone, M.R.; Vignat, J.; Sylla, B.S.; Del Mistro, A.; Franceschi, S.; Tommasino, M.; Clifford, G.M. HPV16 genetic variation and the development of cervical cancer worldwide. Br. J. Cancer 2013, 108, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Gheit, T.; Cornet, I.; Clifford, G.M.; Iftner, T.; Munk, C.; Tommasino, M.; Kjaer, S.K. Risks for persistence and progression by human papillomavirus type 16 variant lineages among a population-based sample of Danish women. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Hildesheim, A.; Schiffman, M.; Bromley, C.; Wacholder, S.; Herrero, R.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Scarpidis, U.; Lin, Q.-Q.; et al. Human papillomavirus type 16 variants and risk of cervical cancer. J. Natl. Cancer Inst. 2001, 93, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Pientong, C.; Wongwarissara, P.; EkalaksanananEmail, T.; Swangphon, P.; Kleebkaow, P.; Kongyingyoes, B.; Siriaunkgul, S.; Tungsinmunkong, K.; Suthipintawong, C. Association of human papillomavirus type 16 long control region mutation and cervical cancer. Virol. J. 2013, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Rabelo-Santos, S.H.; Villa, L.L.; Derchain, S.F.; Ferreira, S.; Sarian, L.O.; Angelo-Andrade, L.A.; do Amaral Westin, M.C.; Zeferino, L.C. Variants of human papillomavirus types 16 and 18: Histological findings in women referred for atypical glandular cells or adenocarcinoma in situ in cervical smear. Int. J. Gynecol. Pathol. 2006, 25, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Rodriguez, A.C.; Chen, Z.; Wacholder, S.; Herrero, R.; Hildesheim, A.; Desalle, R.; Befano, B.; Yu, K.; Safaeian, M.; et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 2010, 70, 3159–3169. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.F.; Koutsky, L.A.; Hildesheim, A.; Galloway, D.A.; Wheeler, C.M.; Winer, R.L.; Ho, J.; Kiviat, N.B. Risk for high-grade cervical intraepithelial neoplasia associated with variants of human papillomavirus types 16 and 18. Cancer Epidemiol. Biomark. Prev. 2007, 16, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Zehbe, I.; Voglino, G.; Delius, H.; Wilander, E.; Tommasino, M. Risk of cervical cancer and geographical variations of human papillomavirus 16 E6 polymorphisms. Lancet 1998, 352, 1441–1442. [Google Scholar] [CrossRef]
- Zuna, R.E.; Moore, W.E.; Shanesmith, R.P.; Dunn, S.T.; Wang, S.S.; Schiffman, M.; Blakey, G.L.; Teel, T. Association of HPV16 E6 variants with diagnostic severity in cervical cytology samples of 354 women in a US population. Int. J. Cancer 2009, 125, 2609–2613. [Google Scholar] [CrossRef] [PubMed]
- Sichero, L.; Ferreira, S.; Trottier, H.; Duarte-Franco, E.; Ferenczy, A.; Franco, E.L.; Villa, L.L. High grade cervical lesions are caused preferentially by non-European variants of HPVs 16 and 18. International J. Cancer 2007, 120, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Berumen, J.; Ordoñez, R.M.; Lazcano, E.; Salmeron, J.; Galvan, S.C.; Estrada, R.A.; Yunes, E.; Garcia-Carranca, A.; Gonzalez-Lira, G.; Madrigal-de la Campa, A.; et al. Asian-American Variants of Human Papillomavirus 16 and Risk for Cervical Cancer: A Case–Control Study. J. Natl. Cancer Inst. 2001, 93, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Freitas, L.B.; Chen, Z.; Muqui, E.F.; Boldrini, N.A.T.; Miranda, A.E.; Spano, L.C.; Burk, R.D. Human Papillomavirus 16 Non-European Variants Are Preferentially Associated with High-Grade Cervical Lesions. PLoS ONE 2014, 9, e100746. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.D.; Terai, M.; Gravitt, P.E.; Brinton, L.A.; Kurman, R.J.; Barnes, W.A.; Greenberg, M.D.; Hadjimichael, O.C.; Fu, L.; McGowan, L.; et al. Distribution of Human Papillomavirus Types 16 and 18 Variants in Squamous Cell Carcinomas and Adenocarcinomas of the Cervix. Cancer Res. 2003, 63, 7215–7220. [Google Scholar] [PubMed]
- Quint, K.D.; de Koning, M.N.; van Doorn, L.J.; Quint, W.G.; Pirog, E.C. HPV genotyping and HPV16 variant analysis in glandular and squamous neoplastic lesions of the uterine cervix. Gynecol. Oncol. 2010, 117, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Nicolás-Párraga, S.; Alemany, L.; de Sanjosé, S.; Bosch, F.X.; Bravo, I.G.; RIS HPV TT and HPV VVAP Study Groups. Differential HPV16 variant distribution in squamous cell carcinoma, adenocarcinoma and adenosquamous cell carcinoma. Int. J. Cancer 2017, 140, 2092–2100. [Google Scholar]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 genetic conservation is critical to carcinogenesis. Cell 2017, 170, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Cullen, M.; Boland, J.F.; Schiffman, M.; Zhang, X.; Wenzensen, N.; Yang, Q.; Chen, Z.; Yu, K.; Mitchell, J.; Roberson, D.; et al. Deep sequencing of HPV16 genomes: A new high-throughput tool for exploring the carcinogenicity and natural history of HPV16 infection. Papillomavirus Res. 2015, 1, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Kinney, W.K.; Cheung, L.C.; Gage, J.C.; Fetterman, B.; Poitras, N.E.; Lorey, T.S.; Wentzensen, N.; Befano, B.; Schussler, J.; et al. Relative Performance of HPV and Cytology Components of Cotesting in Cervical Screening. JNCI J. Natl. Cancer Inst. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Carstensen, B.; Møller, H.; Zappa, M.; Zakelj, M.P.; Lawrence, G.; Hakama, M.; Weiderpass, E. Incidence Trends of Adenocarcinoma of the Cervix in 13 European Countries. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Gien, L.T.; Beauchemin, M.-C.; Thomas, G. Adenocarcinoma: A unique cervical cancer. Gynecol. Oncol. 2010, 116, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Di Bonito, L.; Bergeron, C. Cytological screening of endocervical adenocarcinoma. Ann. Pathol. 2012, 32, e8–e14. [Google Scholar] [CrossRef] [PubMed]
- Davy, M.L.J.; Dodd, T.J.; Luke, C.G.; Roder, D.M. Cervical cancer: Effect of glandular cell type on prognosis, treatment, and survival. Obstet. Gynecol. 2003, 101, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Ault, K.A.; Joura, E.A.; Kjaer, S.K.; Iversen, O.E.; Wheeler, C.M.; Perez, G.; Brown, D.R.; Koutsky, L.A.; Garland, S.M.; Olsson, S.E.; et al. Adenocarcinoma in situ and associated human papillomavirus type distribution observed in two clinical trials of a quadrivalent human papillomavirus vaccine. Int. J. Cancer 2011, 128, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar]
- Litwin, T.; Clarke, M.A.; Dean, M.; Wentzensen, N. Somatic Host Cell Alterations in HPV Carcinogenesis. Viruses 2017, 9, 206. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Yomamoto, Y.; Laury, A.; Wang, X.; Nucci, M.R.; McLaughlin-Drubin, M.E.; Münger, K.; Feldman, S.; McKeon, F.D.; Xian, W.; et al. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 10516–10521. [Google Scholar] [CrossRef] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Boon, S.S.; Banks, L. High-risk human papillomavirus E6 oncoproteins interact with 14-3-3zeta in a PDZ binding motif-dependent manner. J. Virol. 2013, 87, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Boon, S.S.; Tomaića, V.; Thomasa, M.; Robertsb, S.; Banksa, L. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J. Virol. 2015, 89, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Doorbar, J. Human papillomavirus infection and induction of neoplasia: A matter of fitness. Curr. Opin. Virol. 2016, 20, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.; Newcombe, R.G.; Green, A.C.; Kelly, C.; Noel Gill, O.; Hall, G.; Fiander, A.N.; Pirotte, E.; Hibbitts, S.J.; Homer, J.; et al. Human papillomavirus infection is rare in nonmalignant tonsil tissue in the UK: Implications for tonsil cancer precursor lesions. Int. J. Cancer 2014, 135, 2437–2443. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Wentzensen, N. Human papillomavirus infection and the multistage carcinogenesis of cervical cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S. Papillomavirus transcripts and posttranscriptional regulation. Virology 2013, 445, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Schwartz, S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat. Rev. Microbiol. 2013, 11, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Schwartz, S. RNA Binding Proteins that Control Human Papillomavirus Gene Expression. Biomolecules 2015, 5, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Straub, E.; Dreer, M.; Fertey, J.; Iftner, T.; Stubenrauch, F. The viral E8^E2C repressor limits productive replication of human papillomavirus 16. J. Virol. 2014, 88, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Vinokurova, S.; Wentzensen, N.; Kraus, I.; Klaes, R.; Driesch, C.; Melsheimer, P.; Kisseljov, F.; Dürst, M.; Schneider, A.; von Knebel Doeberitz, M.; et al. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 2008, 68, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Ridder, R.; Klaes, R.; Vinokurova, S.; Schaefer, U.; von Knebel Doeberitz, M. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene 2002, 21, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Vinokurova, S.; von Knebel Doeberitz, M. Systematic Review of Genomic Integration Sites of Human Papillomavirus Genomes in Epithelial Dysplasia and Invasive Cancer of the Female Lower Genital Tract. Cancer Res. 2004, 64, 3878–3884. [Google Scholar] [CrossRef] [PubMed]
- Bodelon, C.; Vinokurova, S.; Sampson, J.N.; den Boon, J.A.; Walker, J.L.; Horswill, M.A.; Korthauer, K.; Schiffman, M.; Sherman, M.E.; Zuna, R.E.; et al. Chromosomal copy number alterations and HPV integration in cervical precancer and invasive cancer. Carcinogenesis 2016, 37, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Driesch, C.; Jansen, L.; Runnebaum, I.B.; Dürst, M. Non-random integration of the HPV genome in cervical cancer. PLoS ONE 2012, 7, e39632. [Google Scholar] [CrossRef] [PubMed]
- El Awady, M.K.; Kaplan, J.B.; O’Brien, S.J.; Burk, R.D. Molecular analysis of integrated human papillomavirus 16 sequences in the cervical cancer cell line SiHa. Virology 1987, 159, 389–398. [Google Scholar] [CrossRef]
- Bodelon, C.; Untereiner, M.E.; Machiela, M.J.; Vinokurova, S.; Wentzensen, N. Genomic characterization of viral integration sites in HPV-related cancers. Int. J. Cancer 2016, 139, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Sastre-Garau, X.; Schneider-Maunoury, S.; Labib, A.; Orth, G. Integration of papillomavirus DNA near myc genes in genital carcinomas and its consequences for proto-oncogene expression. J. Virol. 1991, 65, 4534–4538. [Google Scholar] [PubMed]
- Jackson, R.; Rosa, B.A.; Lameiras, S.; Cuninghame, S.; Bernard, J.; Floriano, W.B.; Lambert, P.F.; Nicolas, A.; Zehbe, I. Functional variants of human papillomavirus type 16 demonstrate host genome integration and transcriptional alterations corresponding to their unique cancer epidemiology. BMC Genom. 2016, 17, 851. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.; van Poelgeest, M.I.; van der Hulst, J.M.; Drijfhout, J.W.; Fleuren, G.J.; Melief, C.J.; Kenter, G.; Offringa, R.; van der Burg, S.H. Human Papillomavirus Type 16-Positive Cervical Cancer Is Associated with Impaired CD4+ T-Cell Immunity against Early Antigens E2 and E6. Cancer Res. 2004, 64, 5449–5455. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Terai, M.; Fu, L.; Herrero, R.; DeSalle, R.; Burk, R.D. Diversifying Selection in Human Papillomavirus Type 16 Lineages Based on Complete Genome Analyses. J. Virol. 2005, 79, 7014–7023. [Google Scholar] [CrossRef] [PubMed]
- Roura, E.; Castellsagué, X.; Pawlita, M.; Travier, N.; Waterboer, T.; Margall, N.; Bosch, F.X.; de Sanjosé, S.; Dillner, J.; Gram, I.T.; et al. Smoking as a major risk factor for cervical cancer and pre-cancer: Results from the EPIC cohort. Int. J. Cancer 2014, 135, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Juko-Pecirep, I.; Hammer, J.; Ivansson, E.; Enroth, S.; Gustavsson, I.; Feuk, L.; Magnusson, P.K.; McKay, J.D.; Wilander, E.; et al. Genome-wide Association Study of Susceptibility Loci for Cervical Cancer. J. Natl. Cancer Inst. 2013, 105, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, L.; Hu, Z.; Li, S.; Wang, S.; Liu, J.; Wu, C.; He, L.; Zhou, J.; Li, Z.; et al. A genome-wide association study identifies two new cervical cancer susceptibility loci at 4q12 and 17q12. Nat Genet. 2013, 45, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Ivansson, E.L.; Juko-Pecirep, I.; Erlich, H.A.; Gyllensten, U.B. Pathway-based analysis of genetic susceptibility to cervical cancer in situ: HLA-DPB1 affects risk in Swedish women. Genes Immun. 2011, 12, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Zehbe, I.; Mytilineos, J.; Wikström, I.; Henriksen, R.; Edler, L.; Tommasino, M. Association between human papillomavirus 16 E6 variants and human leukocyte antigen class I polymorphism in cervical cancer of Swedish women. Hum. Immunol. 2003, 64, 538–542. [Google Scholar] [CrossRef]
- Zehbe, I.; Tachezy, R.; Mytilineos, J.; Voglino, G.; Mikyskova, I.; Delius, H.; Marongiu, A.; Gissmann, L.; Wilander, E.; Tommasino, M. Human papillomavirus 16 E6 polymorphisms in cervical lesions from different European populations and their correlation with human leukocyte antigen class II haplotypes. Int. J. Cancer 2001, 94, 711–716. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
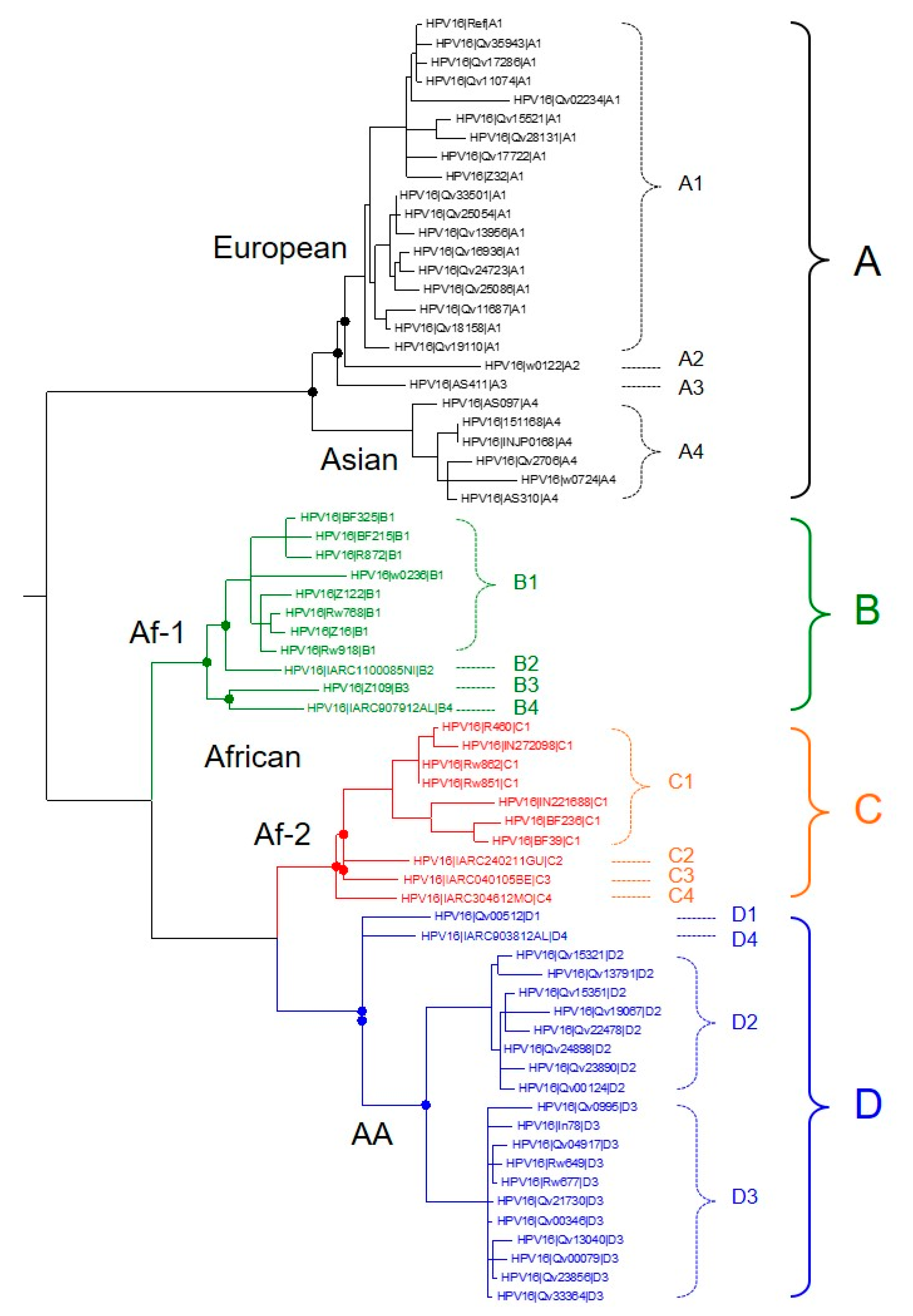{kind=link}
| Topic Area | Questions |
|---|---|
| HPV studies at the species and/or type level and risk of cancer | What features of the biology and/or biochemistry of HPV16 make it so uniquely carcinogenic? |
| What features of HPV16 biology, or interaction with the host cells, enable it to have a wider tissue tropism and disease association? | |
What are the experimental approaches to investigate this?
| |
| Studies of HPV variant lineages within a type to elucidate differences and risk of cancer | Are there functional differences between HPV16 A1 vs. D sublineage viruses that could help explain their pathological differences in cancer risk?
|
| Why are specific HPV16 sublineages (i.e., A4, D2, D3), and HPV18 and HPV45 prone to adenocarcinoma? | |
| What are the next steps after the SNP/gene-based epidemiological approach using case-control datasets? What cell based and/or biochemical experiments could be used to identify the mechanisms of different genetic associations? | |
| Synthesizing current knowledge and moving forward in the era of NGS, systems biology and large data sets | How do we define the characteristics of HPV fitness? |
How do we annotate HPV genomes to be able to capture common functional motifs with disparate genomes in large datasets? (e.g., it’s hard to align a large number of genome sequences of HPV16 with HPV31, HPV52, etc)
| |
| How do we incorporate information on viral suppression/invisibility to the host immune system? | |
| Where does epigenetics of the viral genome fit into the discussion of dissecting viral genome differences? | |
| How to best approach viral—host interactions? |
| Functional Step | Relevant Features |
|---|---|
| 1. Infection | Cell receptor(s) for entry |
| Tissue tropism | |
| 2. Persistence | Continued productive infection |
| Persistence without productive infection | |
| Cellular immunity | |
| Latency | |
| Early, inapparent transformation | |
| 3. Transformation | Increased E6/E7 expression |
| Chromosomal instability | |
| Somatic mutations | |
| Viral integration | |
| 4. Invasion | Increasing somatic mutations |
| Integration, disruption, and partial loss of viral genome | |
| Epithelial-stromal interactions |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirabello, L.; Clarke, M.A.; Nelson, C.W.; Dean, M.; Wentzensen, N.; Yeager, M.; Cullen, M.; Boland, J.F.; NCI HPV Workshop; Schiffman, M.; et al. The Intersection of HPV Epidemiology, Genomics and Mechanistic Studies of HPV-Mediated Carcinogenesis. Viruses 2018, 10, 80. https://doi.org/10.3390/v10020080
Mirabello L, Clarke MA, Nelson CW, Dean M, Wentzensen N, Yeager M, Cullen M, Boland JF, NCI HPV Workshop, Schiffman M, et al. The Intersection of HPV Epidemiology, Genomics and Mechanistic Studies of HPV-Mediated Carcinogenesis. Viruses. 2018; 10(2):80. https://doi.org/10.3390/v10020080
Chicago/Turabian StyleMirabello, Lisa, Megan A. Clarke, Chase W. Nelson, Michael Dean, Nicolas Wentzensen, Meredith Yeager, Michael Cullen, Joseph F. Boland, NCI HPV Workshop, Mark Schiffman, and et al. 2018. "The Intersection of HPV Epidemiology, Genomics and Mechanistic Studies of HPV-Mediated Carcinogenesis" Viruses 10, no. 2: 80. https://doi.org/10.3390/v10020080
APA StyleMirabello, L., Clarke, M. A., Nelson, C. W., Dean, M., Wentzensen, N., Yeager, M., Cullen, M., Boland, J. F., NCI HPV Workshop, Schiffman, M., & Burk, R. D. (2018). The Intersection of HPV Epidemiology, Genomics and Mechanistic Studies of HPV-Mediated Carcinogenesis. Viruses, 10(2), 80. https://doi.org/10.3390/v10020080