High Mobility Group Box 1 Influences HSV1716 Spread and Acts as an Adjuvant to Chemotherapy


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs, Inhibitors Proteins, and Virus
2.3. Cell Secretion Studies
2.4. Virus Quantification
2.5. Cell Proliferation, Viral Spread, and Toxocity Studies
2.6. Conditioned Media
2.7. Western Blot
2.8. shRNA and Lentivirus
2.9. Statistical Analysis
3. Results
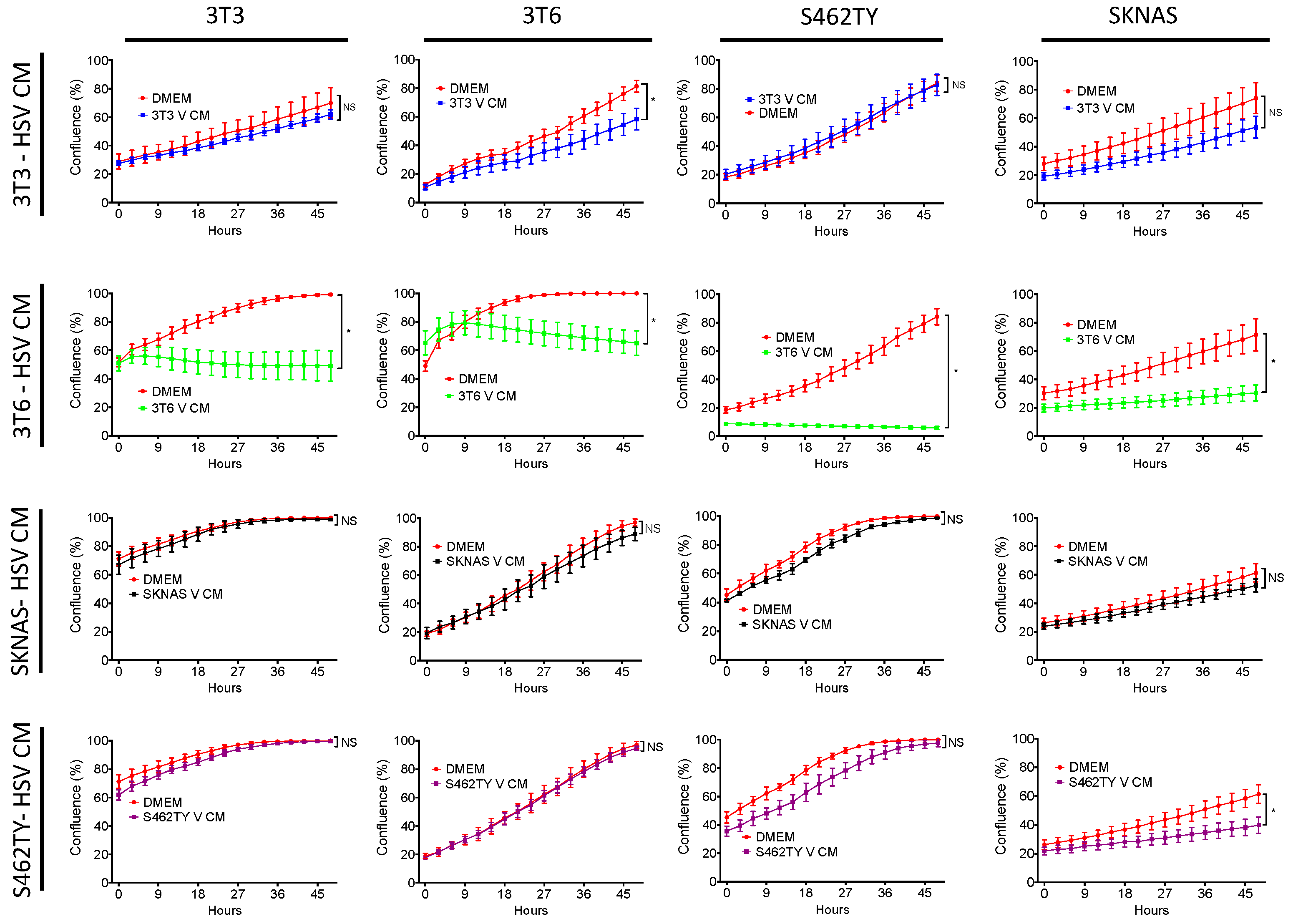
3.1. Conditioned Media from 3T6 Swiss Albino Cells Is Cytotoxic to Bystander Cells
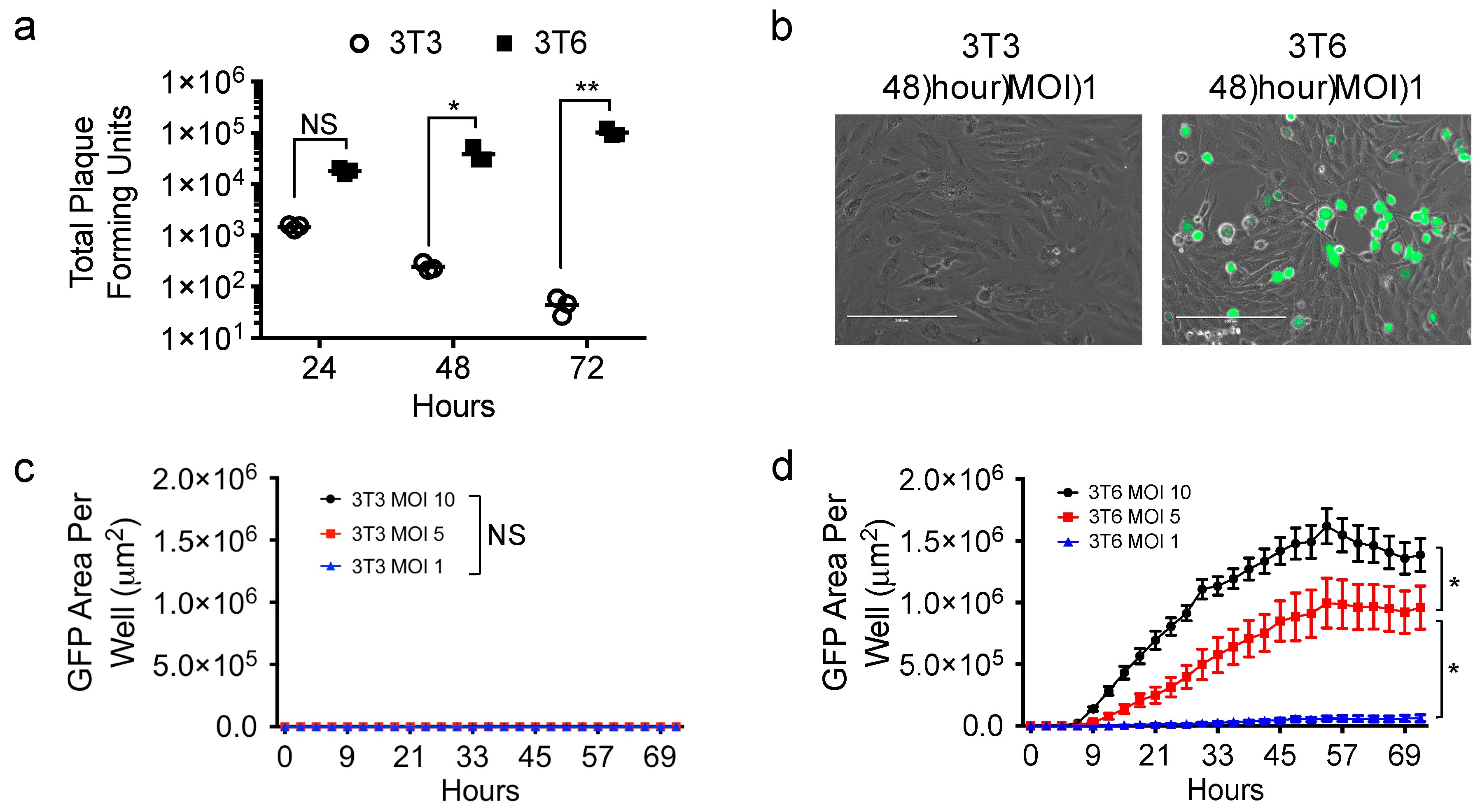
3.2. HSV1716 Infection in NIH-3T3 and 3T6 Swiss Albino
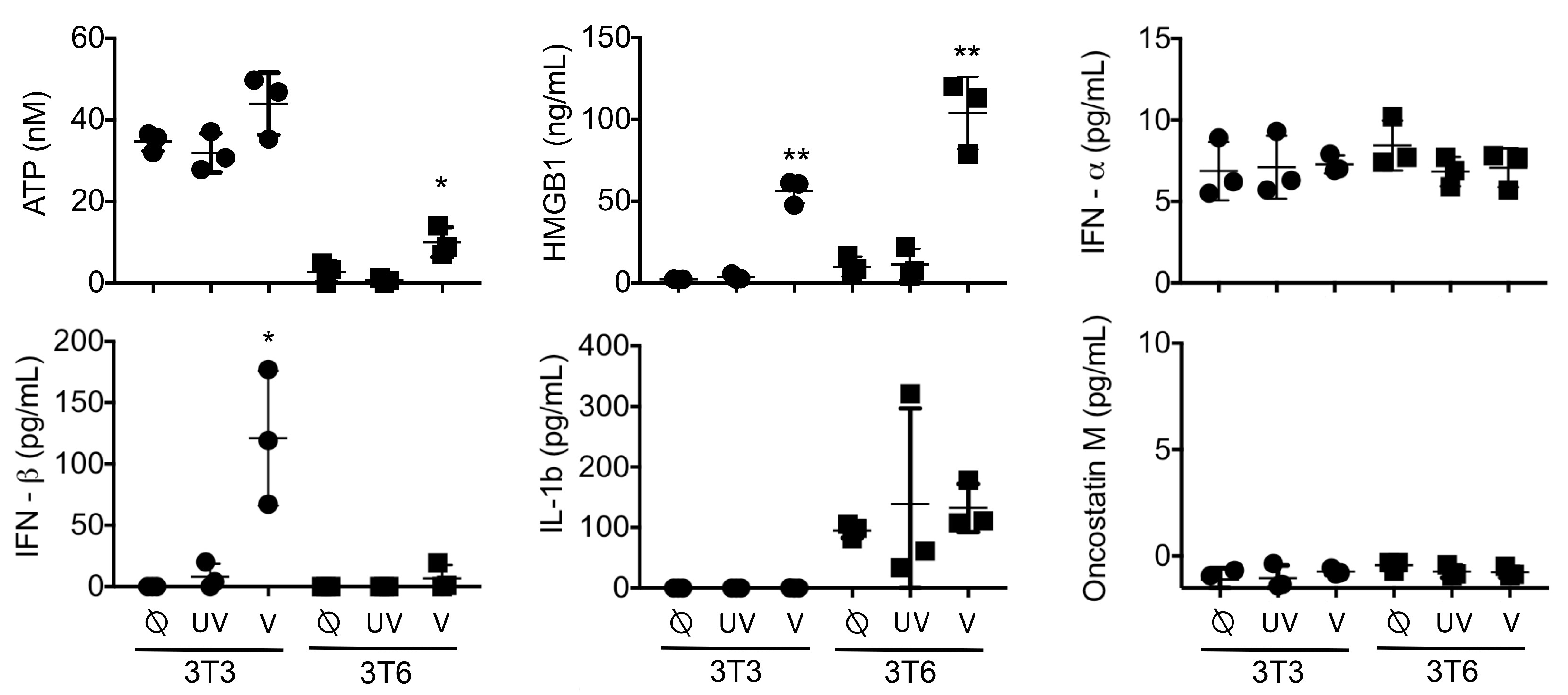
3.3. Active HMGB1 Release Following HSV1716 Infection
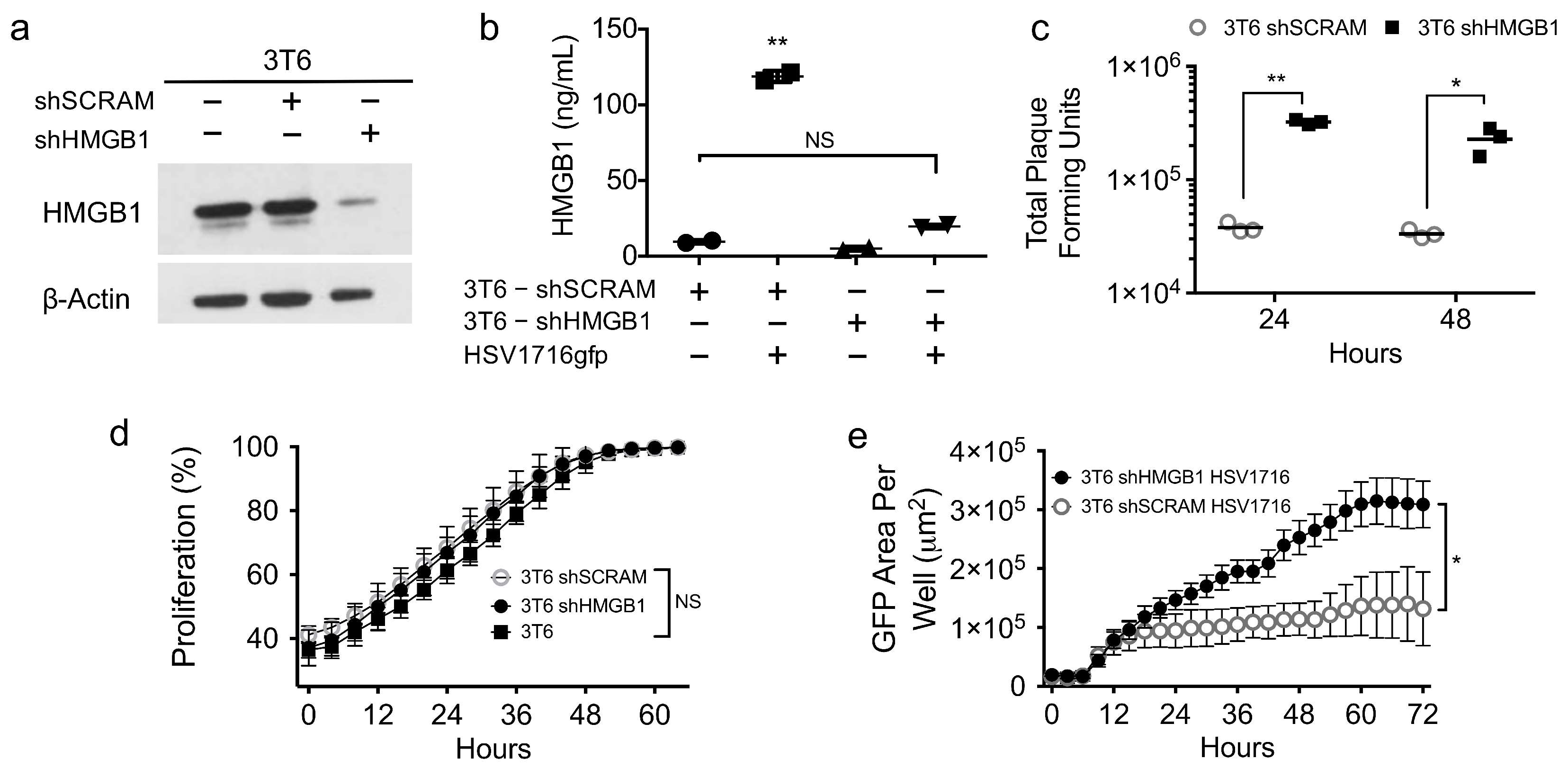
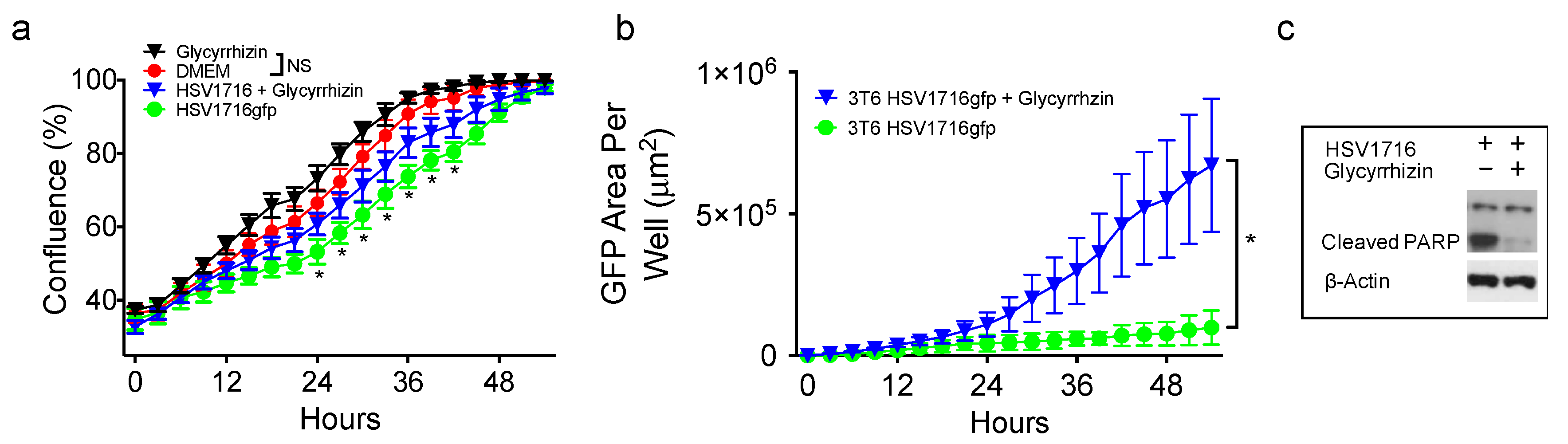
3.4. HMGB1 Inhibition Enhances Virus Spread
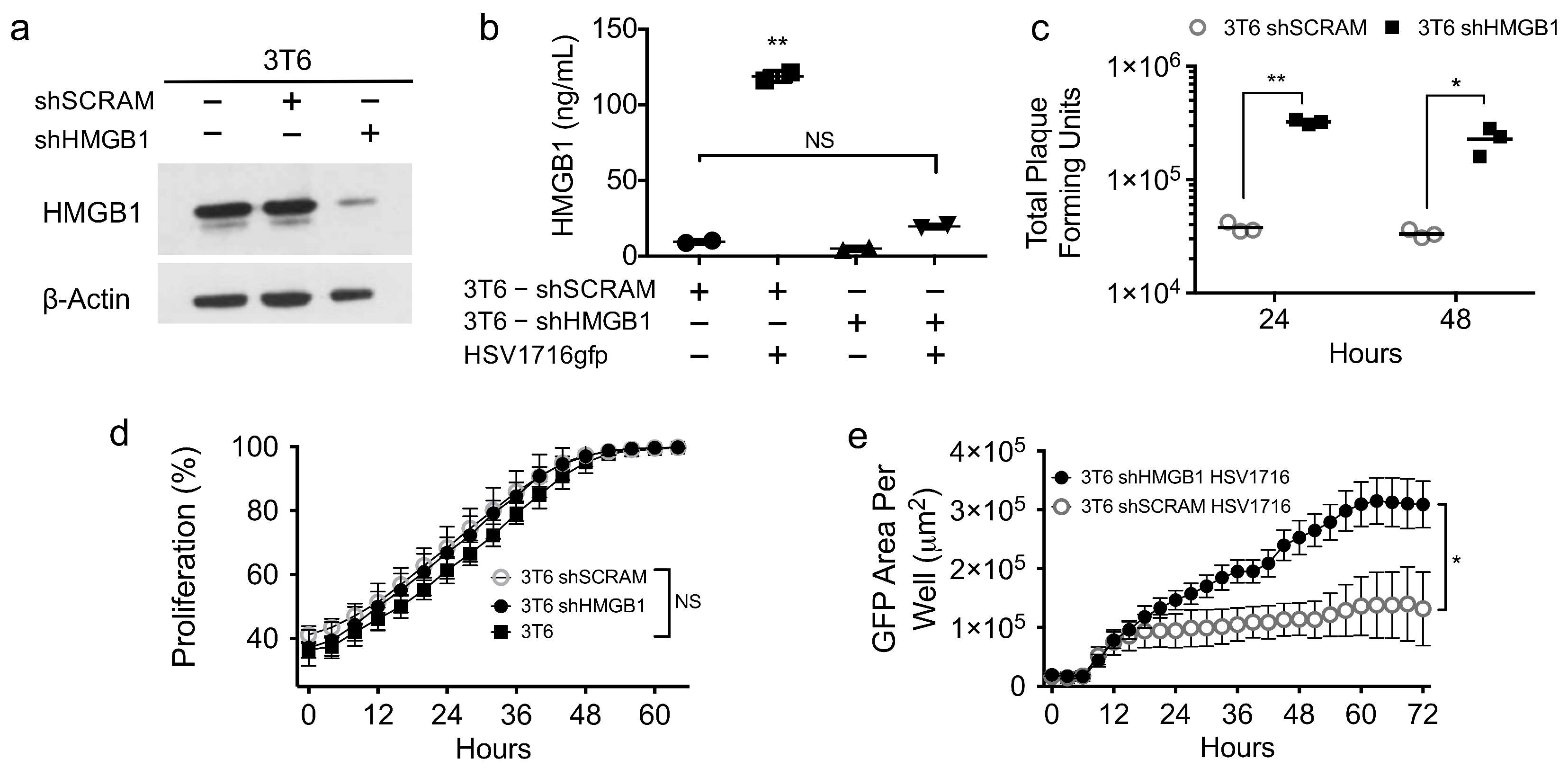
3.5. HMGB1 Knock down Enhances Virus Spread
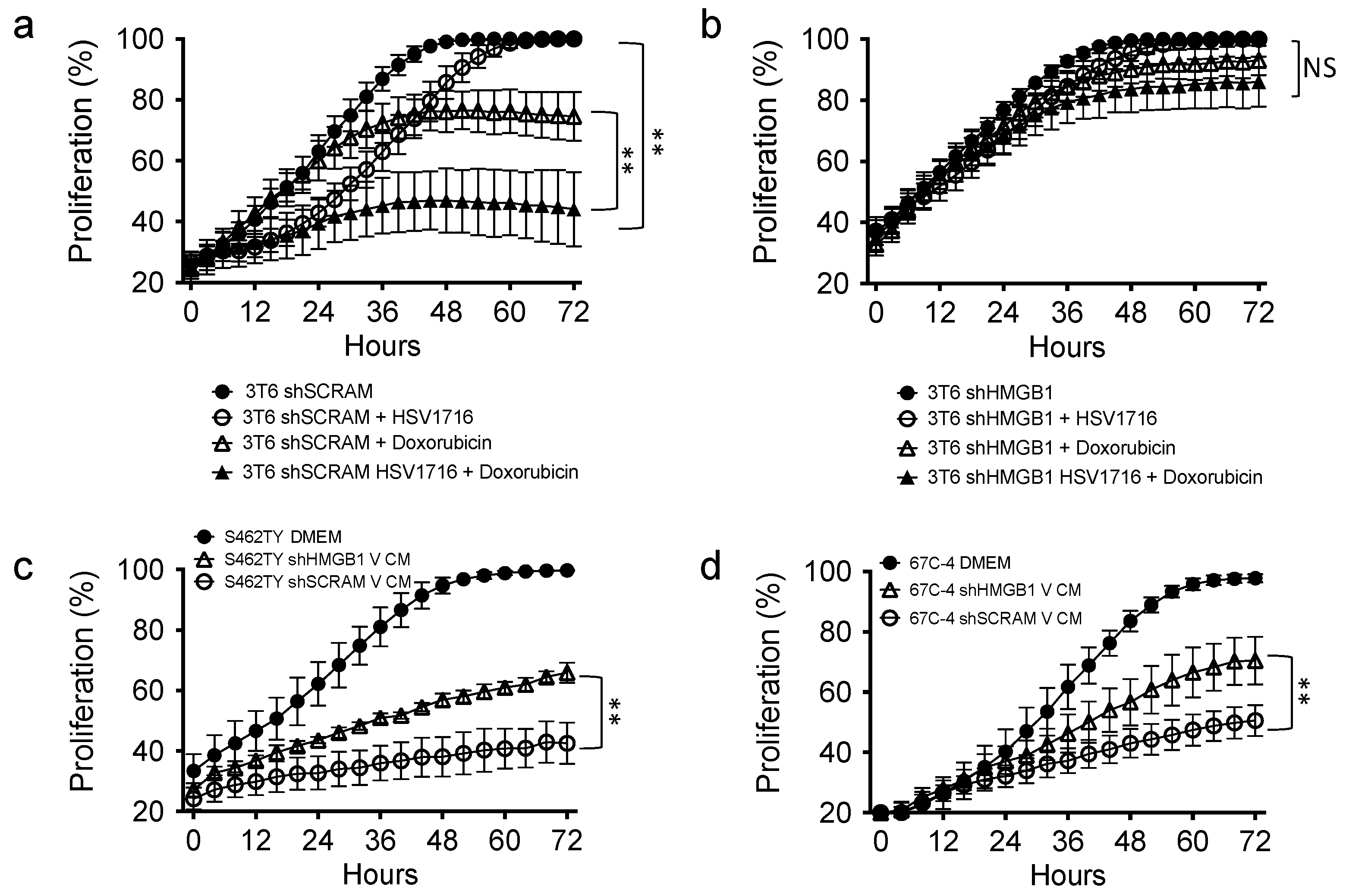
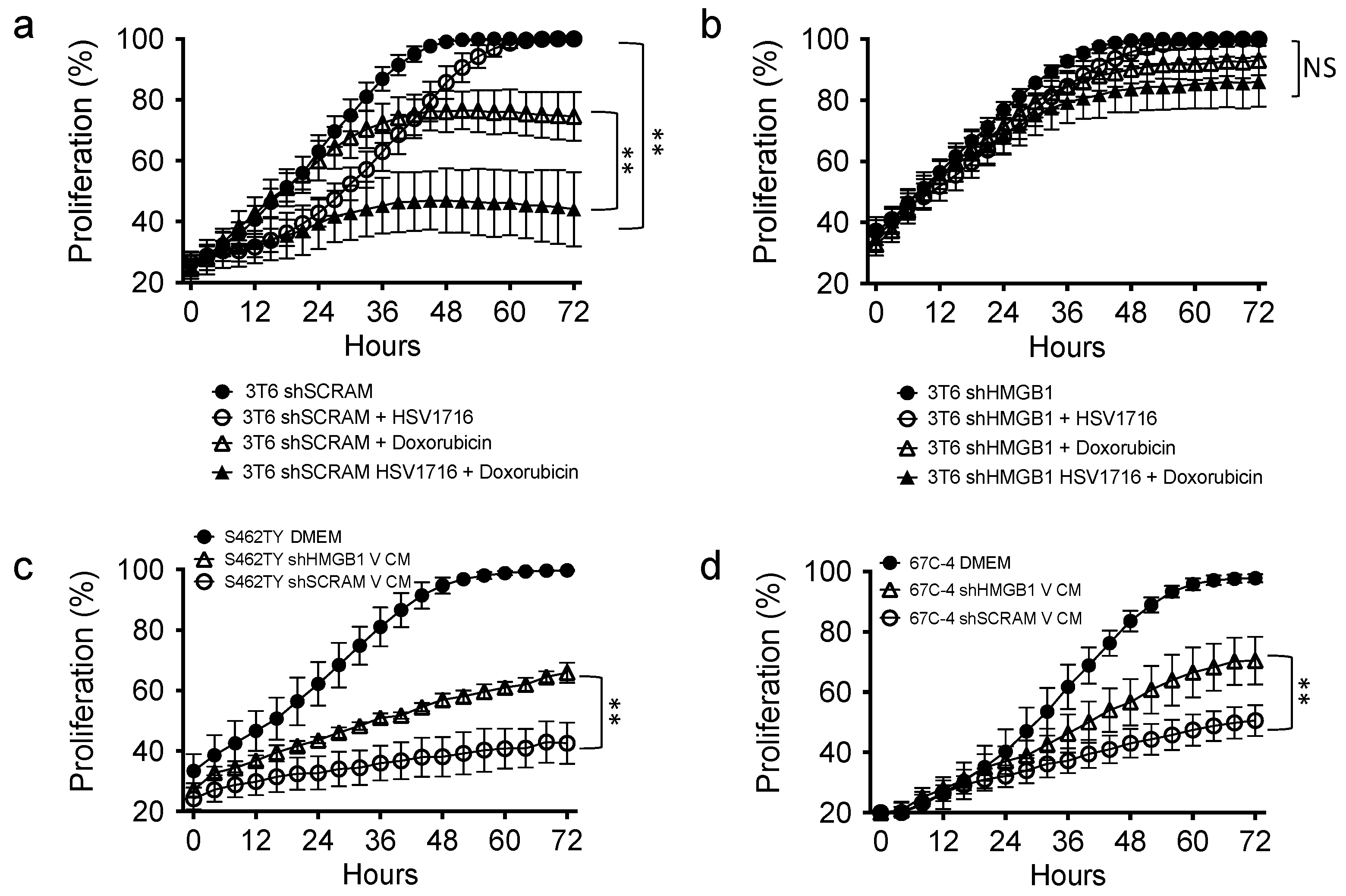
3.6. HMGB1 Influences Drug Sensitivity and Bystander Cell Proliferation
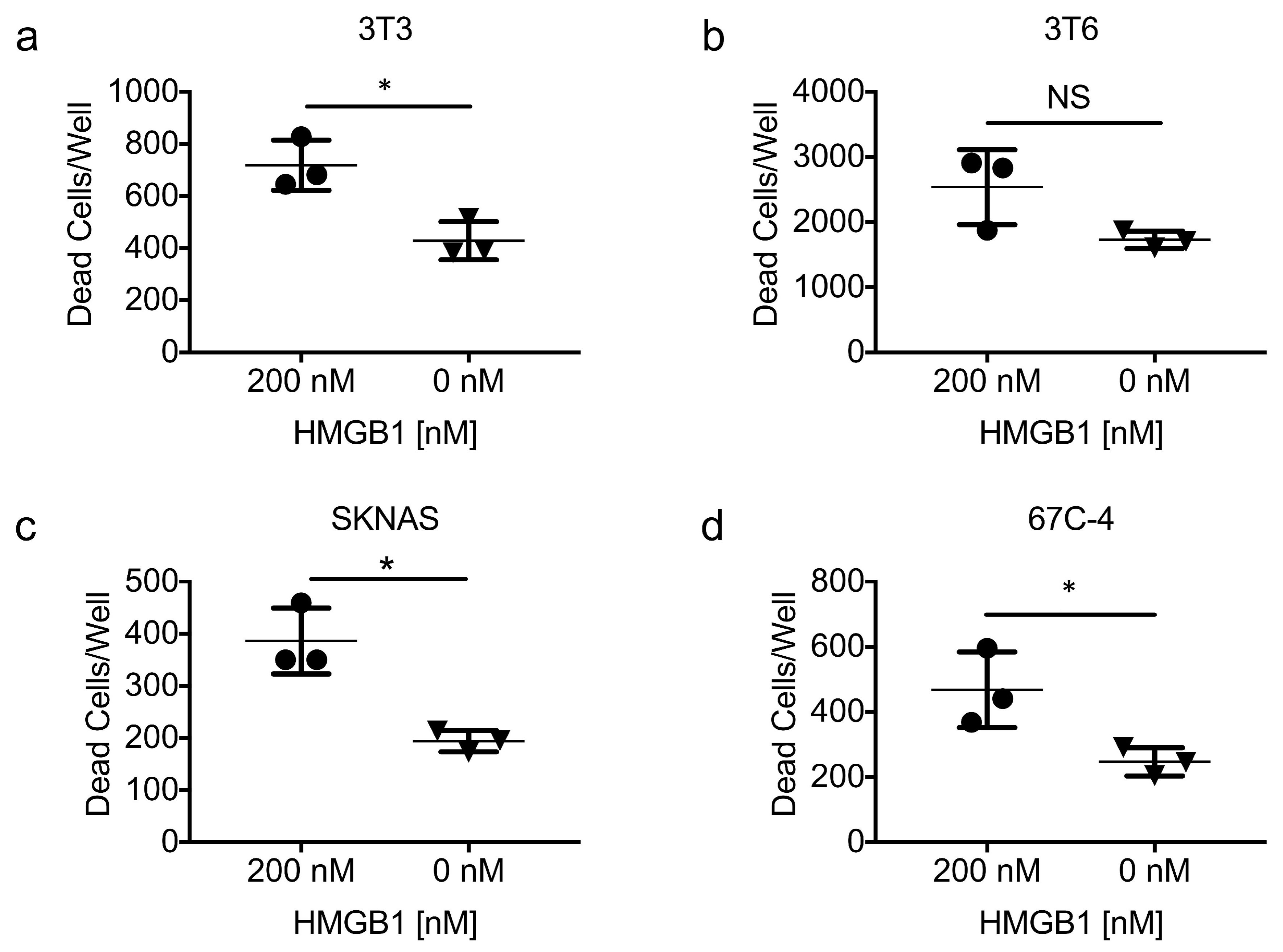
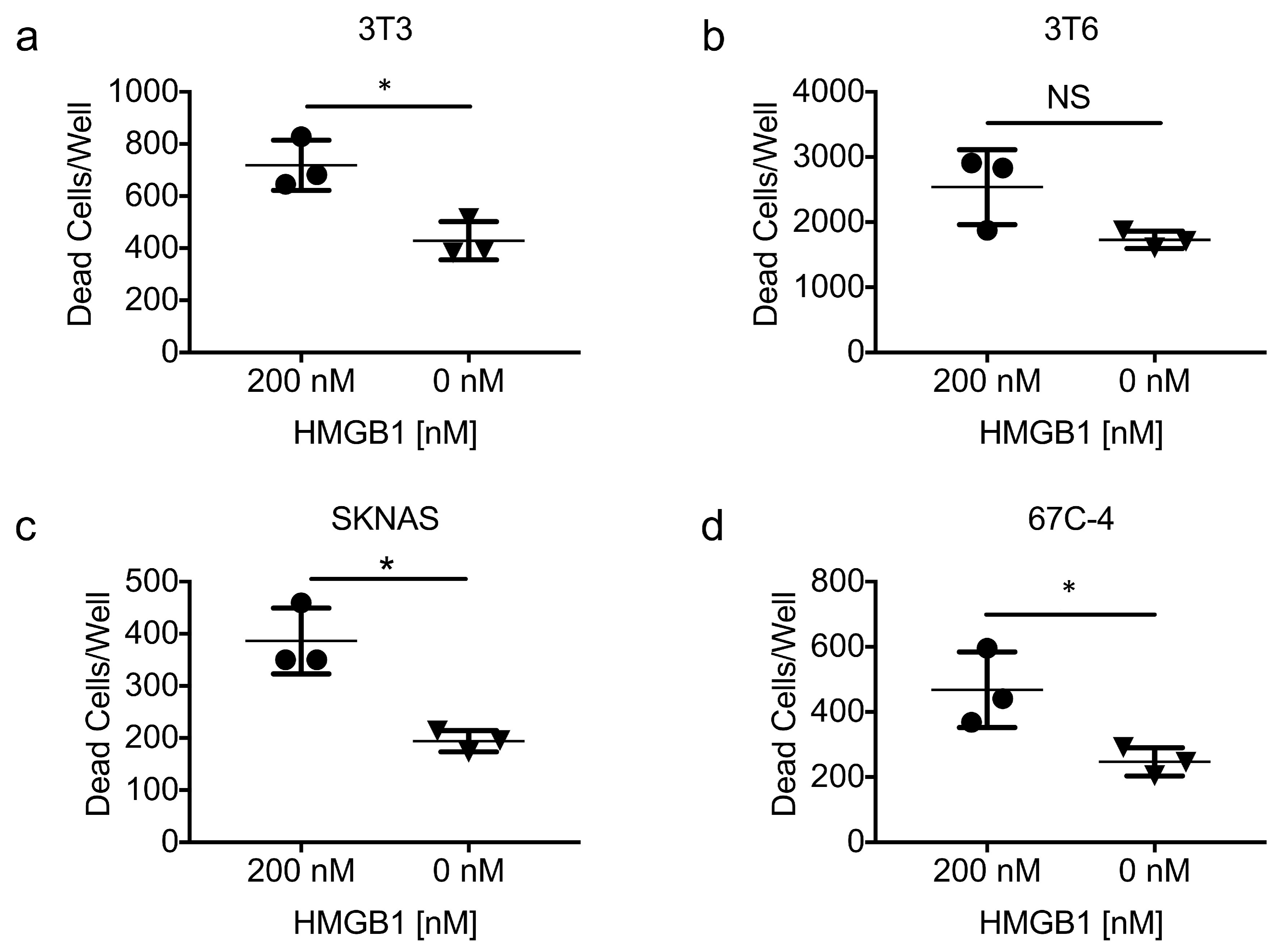
3.7. Exogenous HMGB1 Is Cytotoxic to Multiple Cell Lines
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cassady, K.A.; Haworth, K.B.; Jackson, J.; Markert, J.M.; Cripe, T.P. To infection and beyond: The multi-pronged anti-cancer mechanisms of oncolytic viruses. Viruses 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef] [PubMed]
- McKay, E.M.; McVey, B.; Marsden, H.S.; Brown, S.M.; MacLean, A.R. The herpes simplex virus type 1 strain 17 open reading frame RL1 encodes a polypeptide of apparent m(r) 37k equivalent to ICP34.5 of herpes simplex virus type 1 strain f. J. Gen. Virol. 1993, 74 Pt 11, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- Streby, K.A.; Geller, J.I.; Currier, M.A.; Warren, P.S.; Racadio, J.M.; Towbin, A.J.; Vaughan, M.R.; Triplet, M.; Ott-Napier, K.; Dishman, D.J.; et al. Intratumoral injection of HSV1716, an oncolytic herpes virus, is safe and shows evidence of immune response and viral replication in young cancer patients. Clin. Cancer Res. 2017, 23, 3566–3574. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.; Mairs, R.J.; Braidwood, L.; Joyce, C.; Conner, J.; Pimlott, S.; Brown, M.; Boyd, M. In vivo evaluation of a cancer therapy strategy combining HSV1716-mediated oncolysis with gene transfer and targeted radiotherapy. J. Nucl. Med. 2012, 53, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Currier, M.A.; Sprague, L.; Rizvi, T.A.; Nartker, B.; Chen, C.Y.; Wang, P.Y.; Hutzen, B.J.; Franczek, M.R.; Patel, A.V.; Chaney, K.E.; et al. Aurora a kinase inhibition enhances oncolytic herpes virotherapy through cytotoxic synergy and innate cellular immune modulation. Oncotarget 2017, 8, 17412–17427. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Wang, P.Y.; Hutzen, B.; Sprague, L.; Swain, H.M.; Love, J.K.; Stanek, J.R.; Boon, L.; Conner, J.; Cripe, T.P. Cooperation of oncolytic herpes virotherapy and pd-1 blockade in murine rhabdomyosarcoma models. Sci. Rep. 2017, 7, 2396. [Google Scholar] [CrossRef] [PubMed]
- Hutzen, B.; Chen, C.Y.; Wang, P.Y.; Sprague, L.; Swain, H.M.; Love, J.; Conner, J.; Boon, L.; Cripe, T.P. Tgf-β inhibition improves oncolytic herpes viroimmunotherapy in murine models of rhabdomyosarcoma. Mol. Ther. Oncolytics 2017, 7, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courreges, M.C.; Conejo-Garcia, J.R.; Mohamed-Hadley, A.; Zhang, L.; Buckanovich, R.J.; Carroll, R.; Fraser, N.; Coukos, G. Hsv oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol. Ther. 2005, 12, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courreges, M.C.; Fraser, N.W.; Coukos, G. Herpes virus oncolytic therapy reverses tumor immune dysfunction and facilitates tumor antigen presentation. Cancer Biol. Ther. 2008, 7, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ward, M.F.; Fan, X.G.; Sama, A.E.; Li, W. Potential role of high mobility group box 1 in viral infectious diseases. Viral Immunol. 2006, 19, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic immunotherapy: Dying the right way is a key to eliciting potent antitumor immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaconu, I.; Cerullo, V.; Hirvinen, M.L.; Escutenaire, S.; Ugolini, M.; Pesonen, S.K.; Bramante, S.; Parviainen, S.; Kanerva, A.; Loskog, A.S.; et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012, 72, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, O.G.; Errington-Mais, F.; Steele, L.; Hadac, E.; Jennings, V.; Scott, K.; Peach, H.; Phillips, R.M.; Bond, J.; Pandha, H.; et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 2013, 20, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Borde, C.; Barnay-Verdier, S.; Gaillard, C.; Hocini, H.; Marechal, V.; Gozlan, J. Stepwise release of biologically active HMGB1 during Hsv-2 infection. PLoS ONE 2011, 6, e16145. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Sikorski, R.; Kirn, D.H.; Thorne, S.H. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 2011, 18, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Naik, A.; O’Malley, M.E.; Popovic, P.; Demarco, R.; Hu, Y.; Yin, X.; Yang, S.; Zeh, H.J.; Moss, B.; et al. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005, 65, 9991–9998. [Google Scholar] [CrossRef] [PubMed]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. Hmgb1: Endogenous danger signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Mitchell, D.L.; Vasquez, K.M. High mobility group protein b1 enhances DNA repair and chromatin modification after DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 10320–10325. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Gu, L.; Guo, S.; Wang, C.; Li, G.M. Evidence for involvement of hmgb1 protein in human DNA mismatch repair. J. Biol. Chem. 2004, 279, 20935–20940. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Liu, Y.; Deterding, L.J.; Poltoratsky, V.P.; Kedar, P.S.; Horton, J.K.; Kanno, S.; Asagoshi, K.; Hou, E.W.; Khodyreva, S.N.; et al. HMGB1 is a cofactor in mammalian base excision repair. Mol. Cell 2007, 27, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Vasquez, K.M. HMGB1: The jack-of-all-trades protein is a master DNA repair mechanic. Mol. Carcinog. 2009, 48, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Antoine, D.J.; Kwan, K.; Lundback, P.; Wahamaa, H.; Schierbeck, H.; Robinson, M.; van Zoelen, M.A.; Yang, H.; Li, J.; et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc. Natl. Acad. Sci. USA 2014, 111, 3068–3073. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Ban, T.; Wang, Z.; Choi, M.K.; Kawamura, T.; Negishi, H.; Nakasato, M.; Lu, Y.; Hangai, S.; Koshiba, R.; et al. Hmgb proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 2009, 462, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by hmgb1 and rage. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Zmijewski, J.; Xu, Z.; Abraham, E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J. Immunol. 2008, 180, 2531–2537. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arnandis, I.; Guillen, M.I.; Gomar, F.; Pelletier, J.P.; Martel-Pelletier, J.; Alcaraz, M.J. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1β in osteoarthritic synoviocytes. Arthritis Res. Ther. 2010, 12, R165. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Brasier, A.R.; Casola, A.; Garofalo, R.P.; Kurosky, A. Respiratory syncytial virus infection triggers epithelial HMGB1 release as a damage-associated molecular pattern promoting a monocytic inflammatory response. J. Virol. 2016, 90, 9618–9631. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Simmons, G.; Pol, J.G.; Lichty, B.D.; Halford, W.P.; Mossman, K.L. Immunogenic HSV-mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol. Ther. 2014, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Ottolino-Perry, K.; Diallo, J.S.; Lichty, B.D.; Bell, J.C.; McCart, J.A. Intelligent design: Combination therapy with oncolytic viruses. Mol. Ther. 2010, 18, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Gao, L.; Yeagy, B.; Reid, T. Virus combinations and chemotherapy for the treatment of human cancers. Curr. Opin. Mol. Ther. 2008, 10, 371–379. [Google Scholar] [PubMed]
- Kanai, R.; Wakimoto, H.; Cheema, T.; Rabkin, S.D. Oncolytic herpes simplex virus vectors and chemotherapy: Are combinatorial strategies more effective for cancer? Future Oncol. 2010, 6, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Braidwood, L. Oncolytic HSV1716 in Combination with Targeted Anti Cancer Agents: Identification of Synergistic Interactions and Their Mechanisms of Synergy. Ph.D. Thesis, University of Glasgow, Glasgow, UK, 2016; 196p. [Google Scholar]
- Conner, J.; Rixon, F.J.; Brown, S.M. Herpes simplex virus type 1 strain HSV1716 grown in baby hamster kidney cells has altered tropism for nonpermissive chinese hamster ovary cells compared to HSV1716 grown in vero cells. J. Virol. 2005, 79, 9970–9981. [Google Scholar] [CrossRef] [PubMed]
- Conner, J.; Braidwood, L.; Brown, S.M. A strategy for systemic delivery of the oncolytic herpes virus HSV1716: Redirected tropism by antibody-binding sites incorporated on the virion surface as a glycoprotein d fusion protein. Gene Ther. 2008, 15, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Mahller, Y.Y.; Rangwala, F.; Ratner, N.; Cripe, T.P. Malignant peripheral nerve sheath tumors with high and low RAS-GTP are permissive for oncolytic herpes simplex virus mutants. Pediatr. Blood Cancer 2006, 46, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Saharia, A.; Guittat, L.; Crocker, S.; Lim, A.; Steffen, M.; Kulkarni, S.; Stewart, S.A. Flap endonuclease 1 contributes to telomere stability. Curr. Biol. 2008, 18, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Bolyard, C.; Yoo, J.Y.; Wang, P.Y.; Saini, U.; Rath, K.S.; Cripe, T.P.; Zhang, J.; Selvendiran, K.; Kaur, B. Doxorubicin synergizes with 34.5ENVE to enhance antitumor efficacy against metastatic ovarian cancer. Clin. Cancer Res. 2014, 20, 6479–6494. [Google Scholar] [CrossRef] [PubMed]
- Gdynia, G.; Keith, M.; Kopitz, J.; Bergmann, M.; Fassl, A.; Weber, A.N.; George, J.; Kees, T.; Zentgraf, H.W.; Wiestler, O.D.; et al. Danger signaling protein hmgb1 induces a distinct form of cell death accompanied by formation of giant mitochondria. Cancer Res. 2010, 70, 8558–8568. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Cheh, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. Hmgb1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.C.; Gdynia, G.; Ehemann, V.; Roth, W. The HMGB1 protein sensitizes colon carcinoma cells to cell death triggered by pro-apoptotic agents. Int. J. Oncol. 2015, 46, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.; Cook, A. The cellular tumour antigen p53: Evidence for transformation-related, immunological variants of p53. Virology 1986, 154, 21–30. [Google Scholar] [CrossRef]
- Cuddihy, A.R.; Li, S.; Tam, N.W.; Wong, A.H.; Taya, Y.; Abraham, N.; Bell, J.C.; Koromilas, A.E. Double-stranded-RNA-activated protein kinase PKR enhances transcriptional activation by tumor suppressor p53. Mol. Cell Biol. 1999, 19, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Ishihara, M.; Lamphier, M.S.; Nozawa, H.; Matsuyama, T.; Mak, T.W.; Aizawa, S.; Tokino, T.; Oren, M.; Taniguchi, T. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature 1996, 382, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Fontela, C.; Macip, S.; Martinez-Sobrido, L.; Brown, L.; Ashour, J.; Garcia-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.C.; Parker, J.N.; Shimamura, M.; Cassady, K.A. Spontaneous and engineered compensatory HSV mutants that counteract the host antiviral pkr response. Viruses 2009, 1, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Surget, S.; Khoury, M.P.; Bourdon, J.C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco Targets Ther. 2013, 7, 57–68. [Google Scholar] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.; Dragoi, A.M.; Wang, X.; Dallacosta, C.; Louten, J.; Musco, G.; Sitia, G.; Yap, G.S.; Wan, Y.; Biron, C.A.; et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to cpg-DNA. Blood 2007, 110, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. Hmgb1 in health and disease. Mol. Asp. Med. 2014, 40, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E. Toll-like receptor polymorphisms, inflammatory and infectious diseases, allergies, and cancer. J. Interferon Cytokine Res. 2013, 33, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M.; Canna, S.W.; Slade, K.; Rao, S.; Kreiger, P.A.; Paessler, M.; Kambayashi, T.; Koretzky, G.A. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J. Clin. Investig. 2011, 121, 2264–2277. [Google Scholar] [CrossRef] [PubMed]
- Salaun, B.; Romero, P.; Lebecque, S. Toll-like receptors’ two-edged sword: When immunity meets apoptosis. Eur. J. Immunol. 2007, 37, 3311–3318. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Park, J.H.; Jee, M.H.; Keum, S.J.; Cho, M.S.; Yoon, S.K.; Jang, S.K. Hepatitis c virus infection is blocked by hmgb1 released from virus-infected cells. J. Virol. 2011, 85, 9359–9368. [Google Scholar] [CrossRef] [PubMed]
- Stanziale, S.F.; Petrowsky, H.; Adusumilli, P.S.; Ben-Porat, L.; Gonen, M.; Fong, Y. Infection with oncolytic herpes simplex virus-1 induces apoptosis in neighboring human cancer cells: A potential target to increase anticancer activity. Clin. Cancer Res. 2004, 10, 3225–3232. [Google Scholar] [CrossRef] [PubMed]
- Schaper, F.; Westra, J.; Bijl, M. Recent developments in the role of high-mobility group box 1 in systemic lupus erythematosus. Mol. Med. 2014, 20, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Urbonaviciute, V.; Furnrohr, B.G.; Meister, S.; Munoz, L.; Heyder, P.; De Marchis, F.; Bianchi, M.E.; Kirschning, C.; Wagner, H.; Manfredi, A.A.; et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: Implications for the pathogenesis of sle. J. Exp. Med. 2008, 205, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Polanska, E.; Pospisilova, S.; Stros, M. Binding of histone H1 to DNA is differentially modulated by redox state of HMGB1. PLoS ONE 2014, 9, e89070. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sprague, L.; Lee, J.M.; Hutzen, B.J.; Wang, P.-Y.; Chen, C.-Y.; Conner, J.; Braidwood, L.; Cassady, K.A.; Cripe, T.P. High Mobility Group Box 1 Influences HSV1716 Spread and Acts as an Adjuvant to Chemotherapy. Viruses 2018, 10, 132. https://doi.org/10.3390/v10030132
Sprague L, Lee JM, Hutzen BJ, Wang P-Y, Chen C-Y, Conner J, Braidwood L, Cassady KA, Cripe TP. High Mobility Group Box 1 Influences HSV1716 Spread and Acts as an Adjuvant to Chemotherapy. Viruses. 2018; 10(3):132. https://doi.org/10.3390/v10030132
Chicago/Turabian StyleSprague, Leslee, Joel M. Lee, Brian J. Hutzen, Pin-Yi Wang, Chun-Yu Chen, Joe Conner, Lynne Braidwood, Kevin A. Cassady, and Timothy P. Cripe. 2018. "High Mobility Group Box 1 Influences HSV1716 Spread and Acts as an Adjuvant to Chemotherapy" Viruses 10, no. 3: 132. https://doi.org/10.3390/v10030132