The Host Factor Early Growth Response Gene (EGR-1) Regulates Vaccinia virus Infectivity during Infection of Starved Mouse Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Antibodies and Chemicals
2.2. Virus Infection and Purification
2.3. Virus Infectivity Assays
2.4. Western Blotting
2.4.1. Whole-Cell Lysate Preparation
2.4.2. Electrophoresis and Immunoblotting
2.5. Real Time PCR
2.6. Electron Microscopy
3. Results
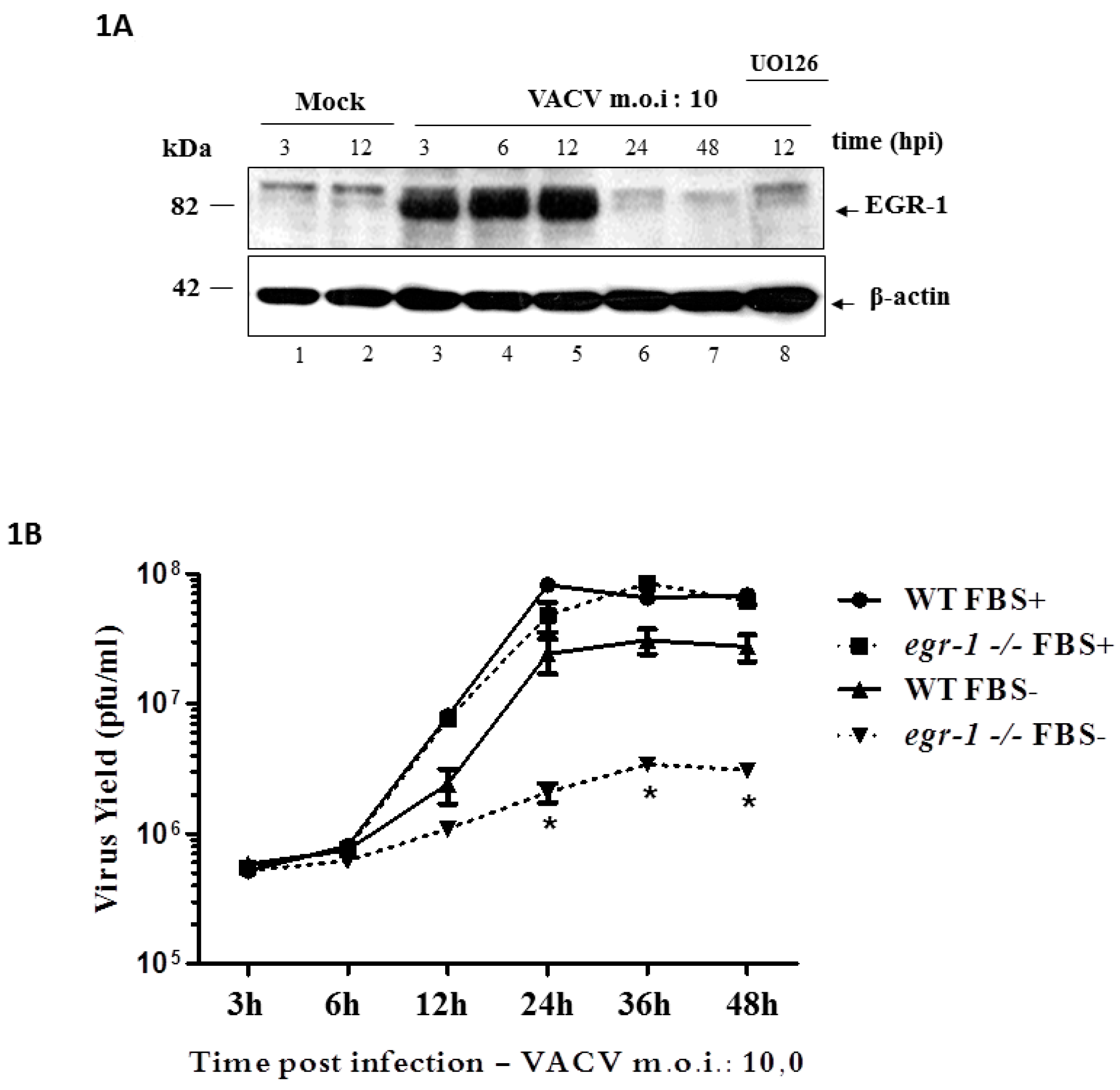
3.1. EGR-1 Protein Expression Is Stimulated through the MEK/ERK Pathway upon VACV Infection and Is Required for Replication in Starved Fibroblasts
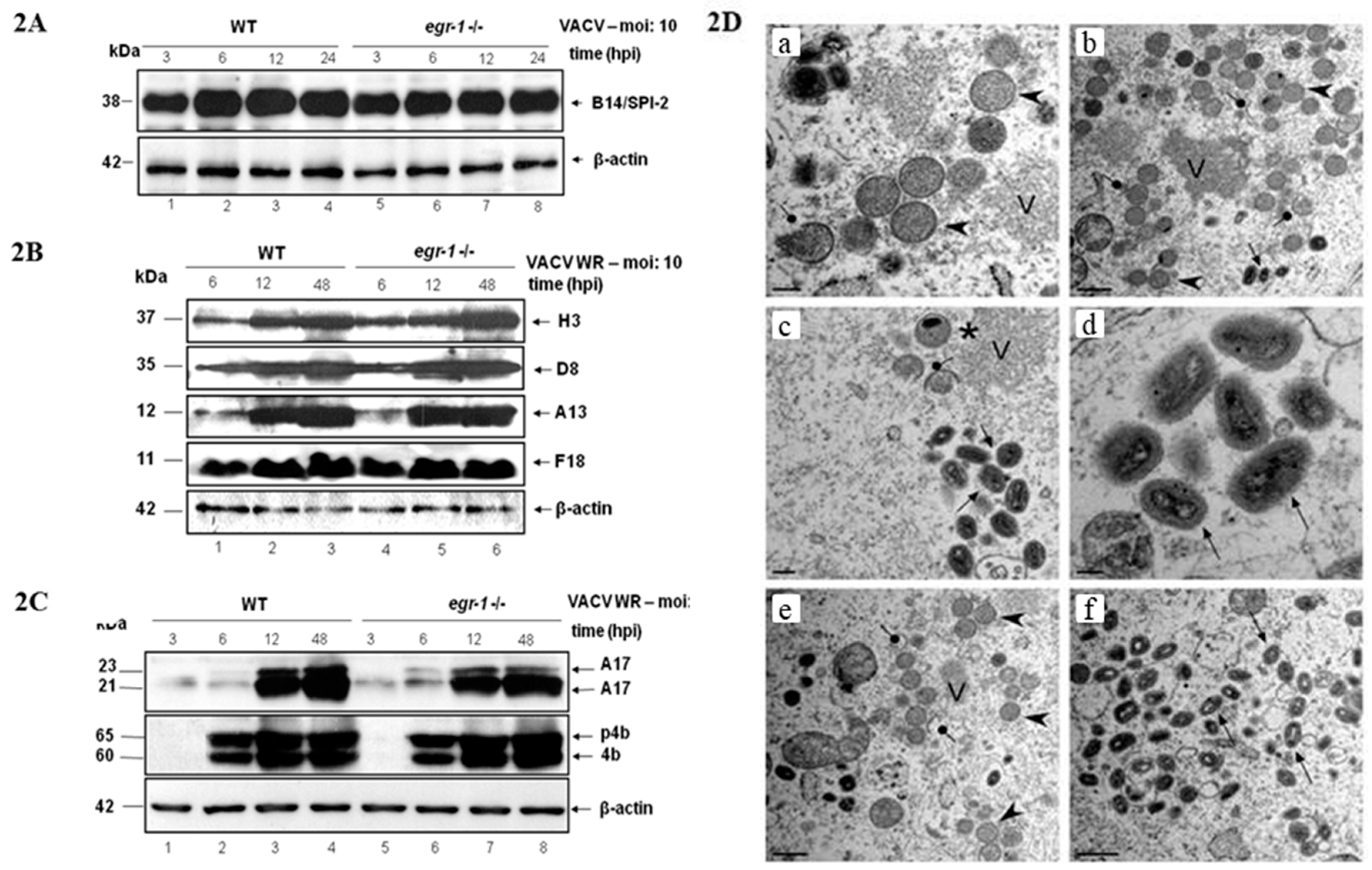
3.2. EGR-1 Gene Deletion Does Not Affect VACV Gene Expression or Morphogenesis in MEFs
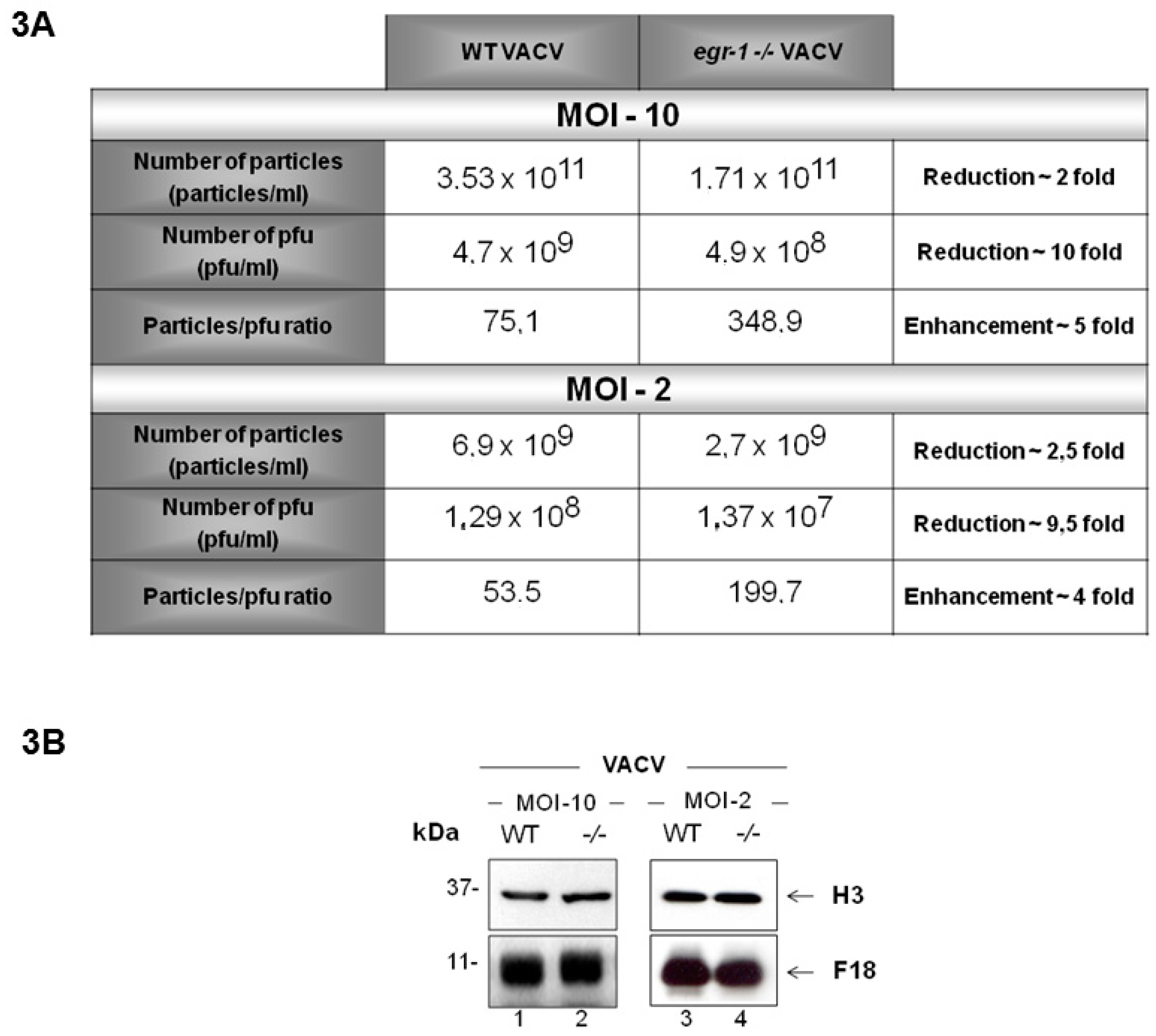
3.3. VACV Produced in egr-1−/− MEFs Have Reduced Specific Infectivity
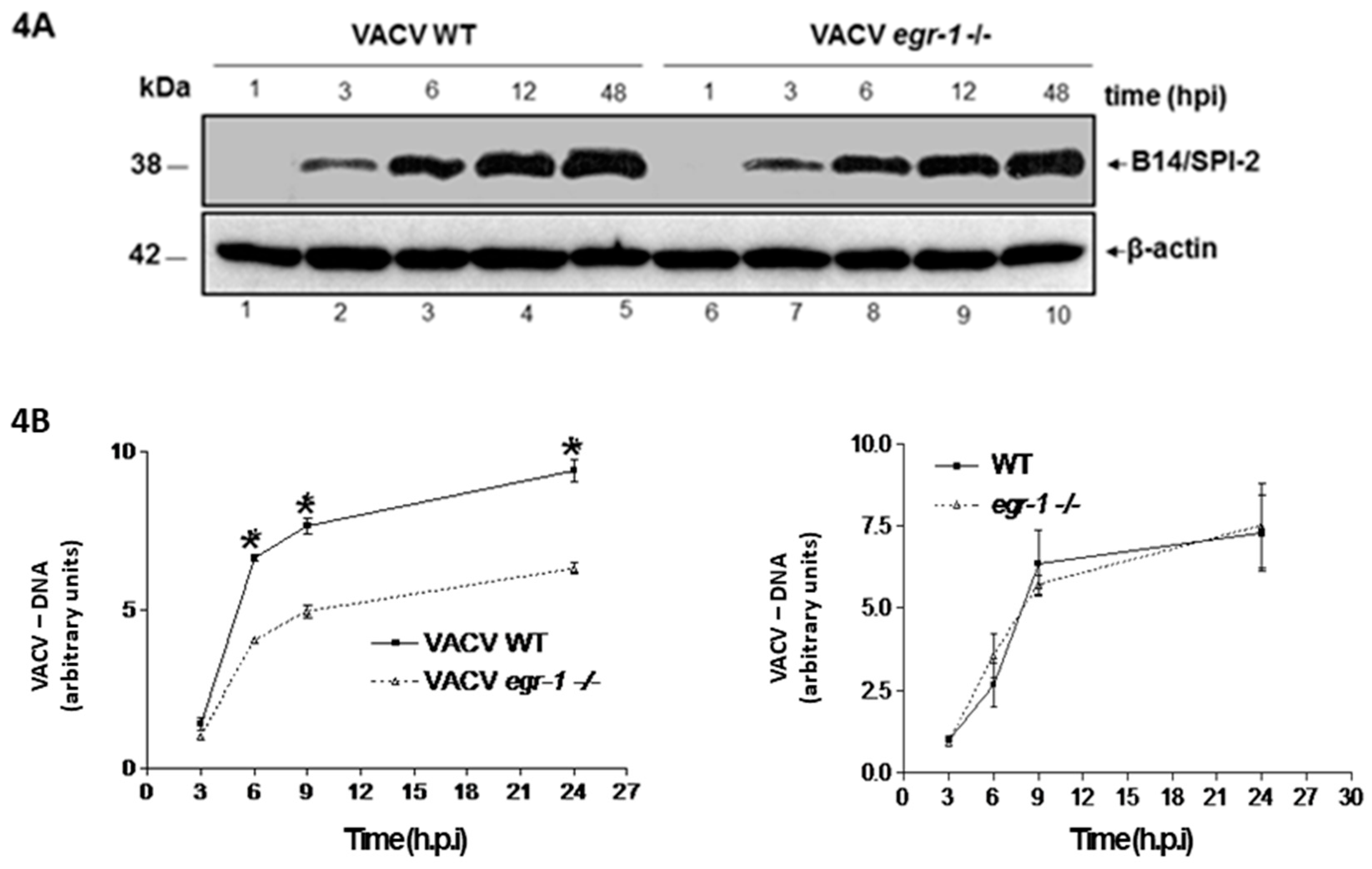
3.4. VACV Produced in MEFs egr-1−/− Presents Delayed Viral DNA Replication during the Next Round of Infection
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Condit, R.C.; Moussatche, N.; Traktman, P. In a nutshell: Structure and assembly of the vaccinia virion. Adv. Virus Res. 2006, 66, 31–124. [Google Scholar] [PubMed]
- Moss, B. Poxviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2130–2174. [Google Scholar]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D. Genetically Engineered Vaccinia Viruses as Agents for Cancer Treatment, Imaging and Transgene Delivery. Front. Oncol. 2017, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.S.; Figueiredo, P.O.; Costa, G.B.; Assis, F.L.; Drumond, B.P.; da Fonseca, F.G.; Nogueira, M.L.; Kroon, E.G.; Trindade, G.S. Vaccinia Virus Natural Infections in Brazil: The Good, the Bad and the Ugly. Viruses 2017, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Wang, C.; Upton, C. Poxviruses: Past, present and future. Virus Res. 2006, 117, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; McFadden, G. Technical knockout: Understanding poxvirus pathogenesis by selectively deleting viral immunomodulatory genes. Cell Microbiol. 2004, 6, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Benfield, C.T.O.; de Motes, C.M.; Mazzon, M.; Ember, S.W.J.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G. Poxvirus tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Bonjardim, C.A. Viral exploitation of the MEK/ERK pathway—A tale of vaccinia virus and other viruses. Virology 2017, 507, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Twardzik, D.R.; Brown, J.P.; Ranchalis, J.E.; Todaro, G.J.; Moss, B. Vaccinia virus-infected cells release a novel polypeptide functionally related to transforming and epidermal growth factors. Proc. Natl. Acad. Sci. USA 1985, 82, 5300–5304. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.P.; Twardzik, D.R.; Marquardt, H.; Todaro, G.J. Vaccinia virus encodes a polypeptide homologous to epidermal growth factor and transforming growth factor. Nature 1985, 313, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Sharrocks, A.D.; Whitmarsh, A.J. MAP kinase signalling cascades and transcriptional regulation. Gene 2013, 513, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, J.C.; Andrade, A.A.; Silva, P.N.; Sousa, L.P.; Ropert, C.; Ferreira, P.C.; Kroon, E.G.; Gazzinelli, R.T.; Bonjardim, C.A. A mitogenic signal triggered at an early stage of vaccinia virus infection: Implication of MEK/ERK and protein kinase A in virus multiplication. J. Biol. Chem. 2001, 276, 38353–38360. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.A.; Silva, P.N.; Pereira, A.C.; de Sousa, L.P.; Ferreira, P.C.; Gazzinelli, R.T.; Kroon, E.G.; Ropert, C.; Bonjardim, C.A. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 2004, 381, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, Y.; Barrett, J.W.; Gao, X.; Loh, J.; Barton, E.; Virgin, H.W.; McFadden, G. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat. Immunol. 2004, 5, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.N.; Soares, J.A.; Brasil, B.S.; Nogueira, S.V.; Andrade, A.A.; de Magalhães, J.C.; Bonjardim, M.B.; Ferreira, P.C.; Kroon, E.G.; Bruna-Romero, O.; et al. Differential role played by the MEK/ERK/EGR-1 pathway in Orthopoxviruses vaccinia and Cowpox biology. Biochem. J. 2006, 398, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Cibelli, G. Regulation of life and death by the zinc finger transcription factor Egr-1. J. Cell. Physiol. 2002, 193, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Rangnekar, V.M.; Adamson, E.; Mercola, D. Suppression of growth and transformation and induction of apoptosis by EGR-1. Cancer Gene Ther. 1998, 5, 3–28. [Google Scholar] [PubMed]
- Sukhatme, V.P.; Cao, X.M.; Chang, L.C.; Tsai-Morris, C.H.; Stamenkovich, D.; Ferreira, P.C.; Cohen, D.R.; Edwards, S.A.; Shows, T.B.; Curran, T. A zinc finger-encoding gene coregulated with c-fos during growth and differentiation and after cellular depolarization. Cell 1988, 53, 37–43. [Google Scholar] [CrossRef]
- Silverman, E.S.; Collins, T. Pathways of Egr-1-mediated gene transcription in vascular biology. Am. J. Pathol. 1999, 154, 665–670. [Google Scholar] [CrossRef]
- Veyrac, A.; Besnard, A.; Caboche, J.; Davis, S.; Laroche, S. The transcription factor Zif268/Egr1, brain plasticity and memory. Prog. Mol. Biol. Transl. Sci. 2014, 122, 89–129. [Google Scholar] [CrossRef] [PubMed]
- Duclot, F.; Kabbaj, M. The Role of Early Growth Response 1 (EGR1) in Brain Plasticity and Neuropsychiatric Disorders. Front. Behav. Neurosci. 2017, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Kim, E.K.; Bae, J.Y.; Moon, S.; Kim, J. Human telomerase reverse transcriptase (hTERT) promotes cancer invasion by modulating cathepsin D via early growth response (EGR)-1. Cancer Lett. 2016, 370, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Dyson, O.F.; Traylen, C.M.; Akula, S.M. Cell Membrane-bound Kaposi’s Sarcoma-associated Herpesvirus-encoded Glycoprotein B Promotes Virus Latency by Regulating Expression of Cellular Egr-1. J. Biol. Chem. 2010, 285, 37491–37502. [Google Scholar] [CrossRef] [PubMed]
- Hsia, S.C.; Graham, L.P.; Bedadala, G.R.; Balish, M.B.; Chen, F.; Figliozzi, R.W. Induction of Transcription Factor Early Growth Response Protein 1 during HSV-1 Infection Promotes Viral Replication in Corneal Cells. Br. Microbiol. Res. J. 2013, 3, 706–723. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, L.; Sariyer, I.K.; Tung, J.; Feliciano, M.; Sawaya, B.E.; Del Valle, L.; Ferrante, P.; Khalili, K.; Safak, M.; White, M.K. Early growth response-1 protein is induced by JC virus infection and binds and regulates the JC virus promoter. Virology 2009, 375, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zou, W.; Green, L.A.; Kim, B.O.; He, J.J. Activation of Egr-1 expression in Astrocytes by HIV-1 Tat: New Insights into Astrocyte-Mediated Tat Neurotoxicity. J. Neuroimmune Pharmacol. 2011, 6, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Vockerodt, M.; Wei, W.; Nagy, E.; Prouzova, Z.; Schrader, A.; Kube, D.; Rowe, M.; Woodman, C.B.; Murray, P.G. Suppression of the LMP2A target gene, EGR-1, protects Hodgkin's lymphoma cells from entry to the EBV lytic cycle. J. Pathol. 2013, 230, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Virolle, T.; Adamson, E.D.; Baron, V.; Birle, D.; Mercola, D.; Mustelin, T.; de Belle, I. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat. Cell Biol. 2001, 3, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Joklik, W.K. The purification of four strains of poxvirus. Virology 1962, 18, 9–18. [Google Scholar] [CrossRef]
- Earl, P.L.; Moss, B.; Wyatt, L.S.; Carroll, M.W. Current Protocols in Molecular Biology; Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Eds.; Wiley: New York, NY, USA, 1998; pp. 118–129. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniats, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; pp. 78–84. [Google Scholar]
- Olson, V.A.; Laue, T.; Laker, M.T.; Babkin, I.V.; Drosten, C.; Shchelkunov, S.N.; Niedrig, M.; Damon, I.K.; Meyer, H. Real-time PCR system for detection of orthopoxviruses and simultaneous identification of smallpox virus. J. Clin. Microbiol. 2004, 42, 1940–1946. [Google Scholar] [CrossRef] [PubMed]
- Broyles, S.S.; Liu, X.; Zhu, M.; Kremer, M. Transcription factor YY1 is a vaccinia virus late promoter activator. J. Biol. Chem. 1999, 274, 35662–35667. [Google Scholar] [CrossRef] [PubMed]
- Katsafanas, G.C.; Moss, B. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J. Biol. Chem. 2004, 279, 52210–52217. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Broyles, S.S. Host cell nuclear proteins are recruited to cytoplasmic vaccinia virus replication complexes. J. Virol. 2005, 79, 12852–12860. [Google Scholar] [CrossRef] [PubMed]
- Knutson, B.A.; Liu, X.; Oh, J.; Broyles, S.S. Vaccinia virus intermediate and late promoter elements are targeted by the TATA-binding protein. J. Virol. 2006, 80, 6784–6793. [Google Scholar] [CrossRef] [PubMed]
- Katsafanas, G.C.; Moss, B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2007, 2, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Simpson-Holley, M.; Kedersha, N.; Dower, K.; Rubins, K.H.; Anderson, P.; Hensley, L.E.; Connor, J.H. Formation of antiviral cytoplasmic granules during Orthopoxvirus infection. J. Virol. 2011, 85, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Burdon, J.J.; Thrall, P.H.; Ericson, L. Genes, communities & invasive species: Understanding the ecological and evolutionary dynamics of host-pathogen interactions. Curr. Opin. Plant. Biol. 2013, 16, 400–405. [Google Scholar] [CrossRef]
- Seet, B.T.; Johnston, J.B.; Brunetti, C.R.; Barrett, J.W.; Everett, H.; Cameron, C.; Sypula, J.; Nazarian, S.H.; Lucas, A.; McFadden, G. Poxviruses and immune evasion. Annu. Rev. Immunol. 2003, 21, 377–423. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.K.; Pyo, C.W.; Kim, Y.; Ahn, B.Y.; Choi, S.Y. Vaccinia virus-mediated cell cycle alteration involves inactivation of tumour suppressors associated with Brf1 and TBP. Cell Microbiol. 2008, 10, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.; Fleming, S.B.; Mercer, A.A. Cell cycle deregulation by a poxvirus partial mimic of anaphase-promoting complex subunit 11. Proc. Natl. Acad. Sci. USA 2009, 106, 19527–19532. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.; Martin, M.C.; Dodding, M.P.; Way, M. Vaccinia-induced EGFR-MEK signalling and the anti-apoptotic protein F1L synergize to suppress cell death during infection. Cell Microbiol. 2009, 11, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar] [PubMed]
- Knutson, B.A.; Oh, J.; Broyles, S.S. Downregulation of vaccinia virus intermediate and late promoters by host transcription factor YY1. J. Gen. Virol. 2009, 90, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Rosales, R.; Harris, N.; Ahn, B.Y.; Moss, B. Purification and identification of a vaccinia virus-encoded intermediate stage promoter-specific transcription factor that has homology to eukaryotic transcription factor SII (TFIIS) and an additional role as a viral RNA polymerase subunit. J. Biol. Chem. 1994, 269, 14260–14267. [Google Scholar] [PubMed]
- Wright, C.F.; Hubbs, A.E.; Gunasinghe, S.K.; Oswald, B.W. A vaccinia virus late transcription factor copurifies with a factor that binds to a viral late promoter and is complemented by extracts from uninfected HeLa cells. J. Virol. 1998, 72, 1446–1451. [Google Scholar] [PubMed]
- Broyles, S.S. Vaccinia virus transcription. J. Gen. Virol. 2003, 84, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.D.; Chen, T.S.; Gagnier, C.R.; Vemulapalli, S.; Maier, C.S.; Hruby, D.E. Pox proteomics: Mass spectrometry analysis and identification of Vaccinia virion proteins. Virol. J. 2006, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.S.; Chen, C.H.; Ho, M.Y.; Huang, C.Y.; Liao, C.L.; Chang, W. Vaccinia virus proteome: Identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 2006, 80, 2127–2140. [Google Scholar] [CrossRef] [PubMed]
- De Silva, F.S.; Moss, B. Effects of vaccinia virus uracil DNA glycosylase catalytic site and deoxyuridinetriphosphatase deletion mutations individually and together on replication in active and quiescent cells and pathogenesis in mice. Virol. J. 2008, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.G.G.; Torres, A.A.; de Oliveira, L.C.; Soares-Martins, J.A.S.; Pereira, A.C.T.C.; Trindade, G.S.; Abrahão, J.S.; Kroon, E.G.; Ferreira, P.C.P.; Bonjardim, C.A. c-Jun integrates signals from both MEK/ERK and MKK/JNK pathways upon vaccinia virus infection. Arch. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxvirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Oliveira, L.C.; Brasil, B.S.A.F.; Unger, B.; Trindade, G.S.; Abrahão, J.S.; Kroon, E.G.; Traktman, P.; Bonjardim, C.A. The Host Factor Early Growth Response Gene (EGR-1) Regulates Vaccinia virus Infectivity during Infection of Starved Mouse Cells. Viruses 2018, 10, 140. https://doi.org/10.3390/v10040140
De Oliveira LC, Brasil BSAF, Unger B, Trindade GS, Abrahão JS, Kroon EG, Traktman P, Bonjardim CA. The Host Factor Early Growth Response Gene (EGR-1) Regulates Vaccinia virus Infectivity during Infection of Starved Mouse Cells. Viruses. 2018; 10(4):140. https://doi.org/10.3390/v10040140
Chicago/Turabian StyleDe Oliveira, Leonardo C., Bruno S. A. F. Brasil, Bethany Unger, Giliane S. Trindade, Jonatas S. Abrahão, Erna G. Kroon, Paula Traktman, and Cláudio A. Bonjardim. 2018. "The Host Factor Early Growth Response Gene (EGR-1) Regulates Vaccinia virus Infectivity during Infection of Starved Mouse Cells" Viruses 10, no. 4: 140. https://doi.org/10.3390/v10040140