Full Genome Sequencing Reveals New Southern African Territories Genotypes Bringing Us Closer to Understanding True Variability of Foot-and-Mouth Disease Virus in Africa

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cells, Viruses and Whole Genome Sequencing
2.2. Bioinformatic Analysis
2.2.1. Analysis of Next Generation Sequencing Reads
2.2.2. RNA Structure Prediction
2.3. Phylogenetic Analysis
2.3.1. Dataset
2.3.2. Multiple Sequence Alignment
2.3.3. Maximum Likelihood Method
2.3.4. Phylogenetic Signal Scan
2.3.5. Divergence Time Estimate
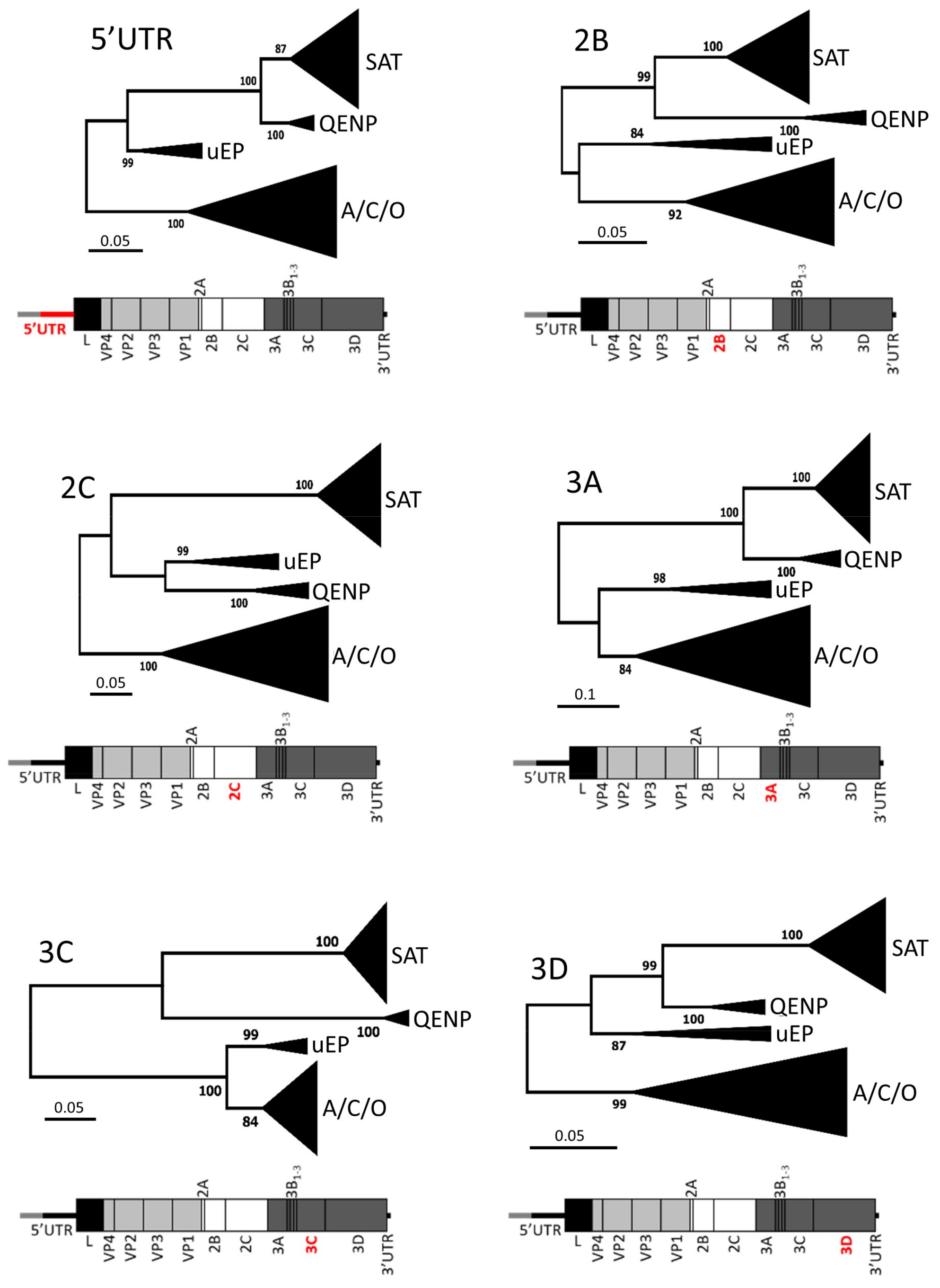
3. Results and Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gloster, J.; Sellers, R.F.; Donaldson, A.I. Long distance transport of foot-and-mouth disease virus over the sea. Vet. Rec. 1982, 110, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Coetzer, J.A.W.; Thomsen, G.R.; Tustin, R.C.; Kriek, N.P.J. (Eds.) Foot-and-mouth disease. In Infectious Diseases of Livestock with Special Reference to Southern Africa; Oxford University Press: Cape Town, South Africa, 1994; pp. 825–852. [Google Scholar]
- Scudamore, J.M.; Harris, D.M. Control of foot and mouth disease: Lessons from the experience of the outbreak in Great Britain in 2001. Rev. Sci. Tech. 2002, 21, 699–710. [Google Scholar] [CrossRef] [PubMed]
- King, D.P.; Henstock, M. Oie/Fao Foot-and-Mouth Disease Reference Laboratory Network Annual Report 2016; The Pirbright Institute: Woking, UK, 2016; pp. 6–17. [Google Scholar]
- Jackson, T.; King, A.M.; Stuart, D.I.; Fry, E. Structure and receptor binding. Virus Res. 2003, 91, 33–46. [Google Scholar] [CrossRef]
- Mason, P.W.; Grubman, M.J.; Baxt, B. Molecular basis of pathogenesis of FMDV. Virus Res. 2003, 91, 9–32. [Google Scholar] [CrossRef]
- Lopez de Quinto, S.; Martinez-Salas, E. Conserved structural motifs located in distal loops of aphthovirus internal ribosome entry site domain 3 are required for internal initiation of translation. J. Virol. 1997, 71, 4171–4175. [Google Scholar] [PubMed]
- Kloc, A.; Diaz-San Segundo, F.; Schafer, E.A.; Rai, D.K.; Kenney, M.; de Los Santos, T.; Rieder, E. Foot-and-mouth disease virus 5′-terminal s fragment is required for replication and modulation of the innate immune response in host cells. Virology 2017, 512, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Belsham, G.J.; Brangwyn, J.K. A region of the 5′ noncoding region of foot-and-mouth disease virus RNA directs efficient internal initiation of protein synthesis within cells: Involvement with the role of l protease in translational control. J. Virol. 1990, 64, 5389–5395. [Google Scholar] [PubMed]
- Mason, P.W.; Bezborodova, S.V.; Henry, T.M. Identification and characterization of a cis-acting replication element (CRE) adjacent to the internal ribosome entry site of foot-and-mouth disease virus. J. Virol. 2002, 76, 9686–9694. [Google Scholar] [CrossRef] [PubMed]
- Serrano, P.; Pulido, M.R.; Saiz, M.; Martinez-Salas, E. The 3′ end of the foot-and-mouth disease virus genome establishes two distinct long-range RNA-RNA interactions with the 5′ end region. J. Gen. Virol. 2006, 87, 3013–3022. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.; Luz, N.; Beck, E. Functional analysis of the internal translation initiation site of foot-and-mouth disease virus. J. Virol. 1990, 64, 4625–4631. [Google Scholar] [PubMed]
- Newton, S.E.; Carroll, A.R.; Campbell, R.O.; Clarke, B.E.; Rowlands, D.J. The sequence of foot-and-mouth disease virus RNA to the 5′ side of the poly(c) tract. Gene 1985, 40, 331–336. [Google Scholar] [CrossRef]
- Brown, F.; Newman, J.; Stott, J.; Porter, A.; Frisby, D.; Newton, C.; Carey, N.; Fellner, P. Poly(c) in animal viral RNAs. Nature 1974, 251, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Vakharia, V.N.; Devaney, M.A.; Moore, D.M.; Dunn, J.J.; Grubman, M.J. Proteolytic processing of foot-and-mouth disease virus polyproteins expressed in a cell-free system from clone-derived transcripts. J. Virol. 1987, 61, 3199–3207. [Google Scholar] [PubMed]
- Klump, W.; Marquardt, O.; Hofschneider, P.H. Biologically active protease of foot and mouth disease virus is expressed from cloned viral cDNA in Escherichia coli. Proc. Natl. Acad. Sci. USA 1984, 81, 3351–3355. [Google Scholar] [CrossRef] [PubMed]
- Strebel, K.; Beck, E. A second protease of foot-and-mouth disease virus. J. Virol. 1986, 58, 893–899. [Google Scholar] [PubMed]
- Belsham, G.J. Translation and replication of FMDV RNA. Curr. Top. Microbiol. Immunol. 2005, 288, 43–70. [Google Scholar] [PubMed]
- Curry, S.; Fry, E.; Blakemore, W.; Abu-Ghazaleh, R.; Jackson, T.; King, A.; Lea, S.; Newman, J.; Stuart, D. Dissecting the roles of vp0 cleavage and RNA packaging in picornavirus capsid stabilization: The structure of empty capsids of foot-and-mouth disease virus. J. Virol. 1997, 71, 9743–9752. [Google Scholar] [PubMed]
- Devaney, M.A.; Vakharia, V.N.; Lloyd, R.E.; Ehrenfeld, E.; Grubman, M.J. Leader protein of foot-and-mouth disease virus is required for cleavage of the p220 component of the cap-binding protein complex. J. Virol. 1988, 62, 4407–4409. [Google Scholar] [PubMed]
- Medina, M.; Domingo, E.; Brangwyn, J.K.; Belsham, G.J. The two species of the foot-and-mouth disease virus leader protein, expressed individually, exhibit the same activities. Virology 1993, 194, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; Morelli, M.J.; Thebaud, G.; Knowles, N.J.; Herzyk, P.; Paton, D.J.; Haydon, D.T.; King, D.P. Beyond the consensus: Dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J. Virol. 2011, 85, 2266–2275. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Escarmis, C.; Baranowski, E.; Ruiz-Jarabo, C.M.; Carrillo, E.; Nunez, J.I.; Sobrino, F. Evolution of foot-and-mouth disease virus. Virus Res. 2003, 91, 47–63. [Google Scholar] [CrossRef]
- Knowles, N.J.; Samuel, A.R. Molecular epidemiology of foot-and-mouth disease virus. Virus Res. 2003, 91, 65–80. [Google Scholar] [CrossRef]
- Rweyemamu, M.; Roeder, P.; Mackay, D.; Sumption, K.; Brownlie, J.; Leforban, Y.; Valarcher, J.F.; Knowles, N.J.; Saraiva, V. Epidemiological patterns of foot-and-mouth disease worldwide. Transbound. Emerg. Dis. 2008, 55, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Samuel, A.R.; Knowles, N.J. Foot-and-mouth disease type o viruses exhibit genetically and geographically distinct evolutionary lineages (topotypes). J. Gen. Virol. 2001, 82, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Hussain, M.; Ahmad, M.; Normann, P.; Afzal, M.; Alexandersen, S. Genetic characterisation of the recent foot-and-mouth disease virus subtype a/irn/2005. Virol. J. 2007, 4, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.N.; Oem, J.K.; Park, J.H.; Kim, S.M.; Lee, S.Y.; Tserendorj, S.; Sodnomdarjaa, R.; Joo, Y.S.; Kim, H. Evidence of recombination in a new isolate of foot-and-mouth disease virus serotype Asia. Virus Res. 2009, 139, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, C.; Tulman, E.R.; Delhon, G.; Lu, Z.; Carreno, A.; Vagnozzi, A.; Kutish, G.F.; Rock, D.L. Comparative genomics of foot-and-mouth disease virus. J. Virol. 2005, 79, 6487–6504. [Google Scholar] [CrossRef] [PubMed]
- Bachanek-Bankowska, K.; di Nardo, A.; Wadsworth, J.; Mioulet, V.; Pezzoni, G.; Grazioli, S.; Brocchi, E.; Kafle, S.C.; Hettiarachchi, R.; Kumarawadu, P.L.; et al. Reconstructing the evolutionary history of pandemic o/me-sa/ind-2001 foot-and-mouth disease viruses. Sci. Rep. 2018. submitted. [Google Scholar]
- Oberste, M.S.; Maher, K.; Pallansch, M.A. Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes. J. Virol. 2004, 78, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; O’Neill, H.; Maree, F.; Blignaut, B.; Carrillo, C.; Rodriguez, L.; Haydon, D.T. Mosaic structure of foot-and-mouth disease virus genomes. J. Gen. Virol. 2007, 88, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Recombination and selection in the evolution of picornaviruses and other mammalian positive-stranded RNA viruses. J. Virol. 2006, 80, 11124–11140. [Google Scholar] [CrossRef] [PubMed]
- Heath, L.; van der Walt, E.; Varsani, A.; Martin, D.P. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef] [PubMed]
- Condy, J.B.; Hedger, R.S.; Hamblin, C.; Barnett, I.T. The duration of the foot-and-mouth disease virus carrier state in African buffalo (i) in the individual animal and (ii) in a free-living herd. Comp. Immunol. Microbiol. Infect. Dis. 1985, 8, 259–265. [Google Scholar] [CrossRef]
- Vosloo, W.; Dwarka, R.M.; Bastos, A.D.S.; Esterhuysen, J.J.; Sahle, M.; Sangare, O. Molecular Epidemiological Studies of Foot-and-Mouth Disease Virus in Sub-Saharan Africa Indicate the Presence of Large Numbers of Topotypes: Implications for Local and International Control; Food and Agriculture Organisation of the United Nations: Chania, Greece, 2004. [Google Scholar]
- Valdazo-Gonzalez, B.; Knowles, N.J.; Hammond, J.; King, D.P. Genome sequences of sat 2 foot-and-mouth disease viruses from Egypt and Palestinian autonomous territories (gaza strip). J. Virol. 2012, 86, 8901–8902. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.A.; Salem, S.A.; Habashi, A.R.; Arafa, A.A.; Aggour, M.G.; Salem, G.H.; Gaber, A.S.; Selem, O.; Abdelkader, S.H.; Knowles, N.J.; et al. Emergence of foot-and-mouth disease virus sat 2 in Egypt during 2012. Transbound. Emerg. Dis. 2012, 59, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, A.; Knowles, N.J.; Paton, D.J. Combining livestock trade patterns with phylogenetics to help understand the spread of foot and mouth disease in sub-saharan Africa, the middle East and Southeast Asia. Rev. Sci. Tech. 2011, 30, 63–85. [Google Scholar] [CrossRef] [PubMed]
- Logan, G.; Freimanis, G.L.; King, D.J.; Valdazo-Gonzalez, B.; Bachanek-Bankowska, K.; Sanderson, N.D.; Knowles, N.J.; King, D.P.; Cottam, E.M. A universal protocol to generate consensus level genome sequences for foot-and-mouth disease virus and other positive-sense polyadenylated RNA viruses using the illumina miseq. BMC Genom. 2014, 15, 828. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, A.; Andino, R. Library preparation for highly accurate population sequencing of RNA viruses. Nat. Protoc. 2014, 9, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for Fastq Files. Version 1.33. 2011. Available online: https://github.com/najoshi/sickle (accessed on 9 February 2011).
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. Idba-ud: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with mafft. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar] [PubMed]
- Katoh, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Improved accuracy of multiple ncrna alignment by incorporating structural information into a mafft-based framework. BMC Bioinform. 2008, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. Rnaalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Honer Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. Viennarna package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. Emboss: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Hurvich, C.M.; Tsai, C.L. Regression and time-series model selection in small samples. Biometrika 1989, 76, 297–307. [Google Scholar] [CrossRef]
- Sugiura, N. Further analysis of data by akaikes information criterion and finite corrections. Commun. Stat.-Theory Methods 1978, 7, 13–26. [Google Scholar] [CrossRef]
- Simmonds, P. SSE: A nucleotide and amino acid sequence analysis platform. BMC Res. Notes 2012, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using tempest (formerly path-o-gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partitionfinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinform. 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, P.; Puller, V.; Neher, R.A. Treetime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Dhikusooka, M.T.; Tjornehoj, K.; Ayebazibwe, C.; Namatovu, A.; Ruhweza, S.; Siegismund, H.R.; Wekesa, S.N.; Normann, P.; Belsham, G.J. Foot-and-mouth disease virus serotype sat 3 in long-horned ankole calf, Uganda. Emerg. Infect. Dis. 2015, 21, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdazo-Gonzalez, B.; Timina, A.; Scherbakov, A.; Abdul-Hamid, N.F.; Knowles, N.J.; King, D.P. Multiple introductions of serotype O foot-and-mouth disease viruses into east Asia in 2010–2011. Vet. Res. 2013, 44, 76. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.D.; Haydon, D.T.; Sangare, O.; Boshoff, C.I.; Edrich, J.L.; Thomson, G.R. The implications of virus diversity within the sat 2 serotype for control of foot-and-mouth disease in sub-saharan Africa. J. Gen. Virol. 2003, 84, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Knowles, N.J.; Wadsworth, J.; Rambaut, A.; Woolhouse, M.E. Reconstructing geographical movements and host species transitions of foot-and-mouth disease virus serotype sat 2. MBio 2013, 4, e00591–e00613. [Google Scholar] [CrossRef] [PubMed]
- Spinage, C.A. Cattle Plague a History; Kluwer Academic/Plenum Publishers: New York, NY, USA; Boston, MA, USA; Dordrecht, The Netherlands; London, UK; Moscow, Russia, 2003. [Google Scholar]
- Casey, M.B.; Lembo, T.; Knowles, N.J.; Fyumagwa, R.; Kivaria, F.; Maliti, H.; Kasanga, C.; Sallu, R.; Reeve, R.; Parida, S.; et al. Chapter 2—Patterns of foot-and-mouth disease virus distribution in Africa: The role of livestock and wildlife in virus emergence. In The Role of Animals in Emerging Viral Diseases; Johnson, N., Ed.; Academic Press: Boston, MA, USA, 2014; pp. 21–38. [Google Scholar]
- Herod, M.R.; Gold, S.; Lasecka-Dykes, L.; Wright, C.; Ward, J.C.; McLean, T.C.; Forrest, S.; Jackson, T.; Tuthill, T.J.; Rowlands, D.J.; et al. Genetic economy in picornaviruses: Foot-and-mouth disease virus replication exploits alternative precursor cleavage pathways. PLoS Pathog. 2017, 13, e1006666. [Google Scholar] [CrossRef] [PubMed]

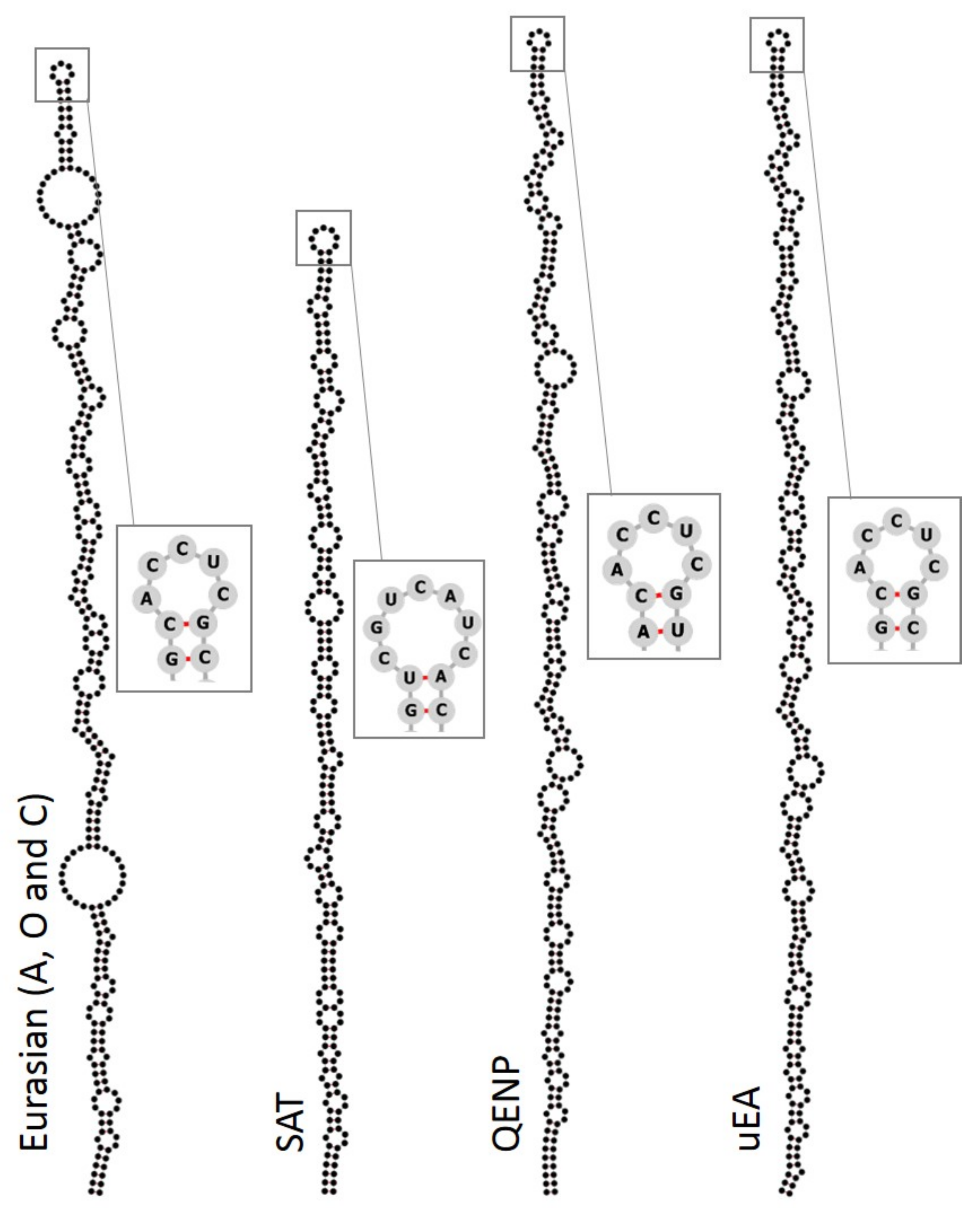

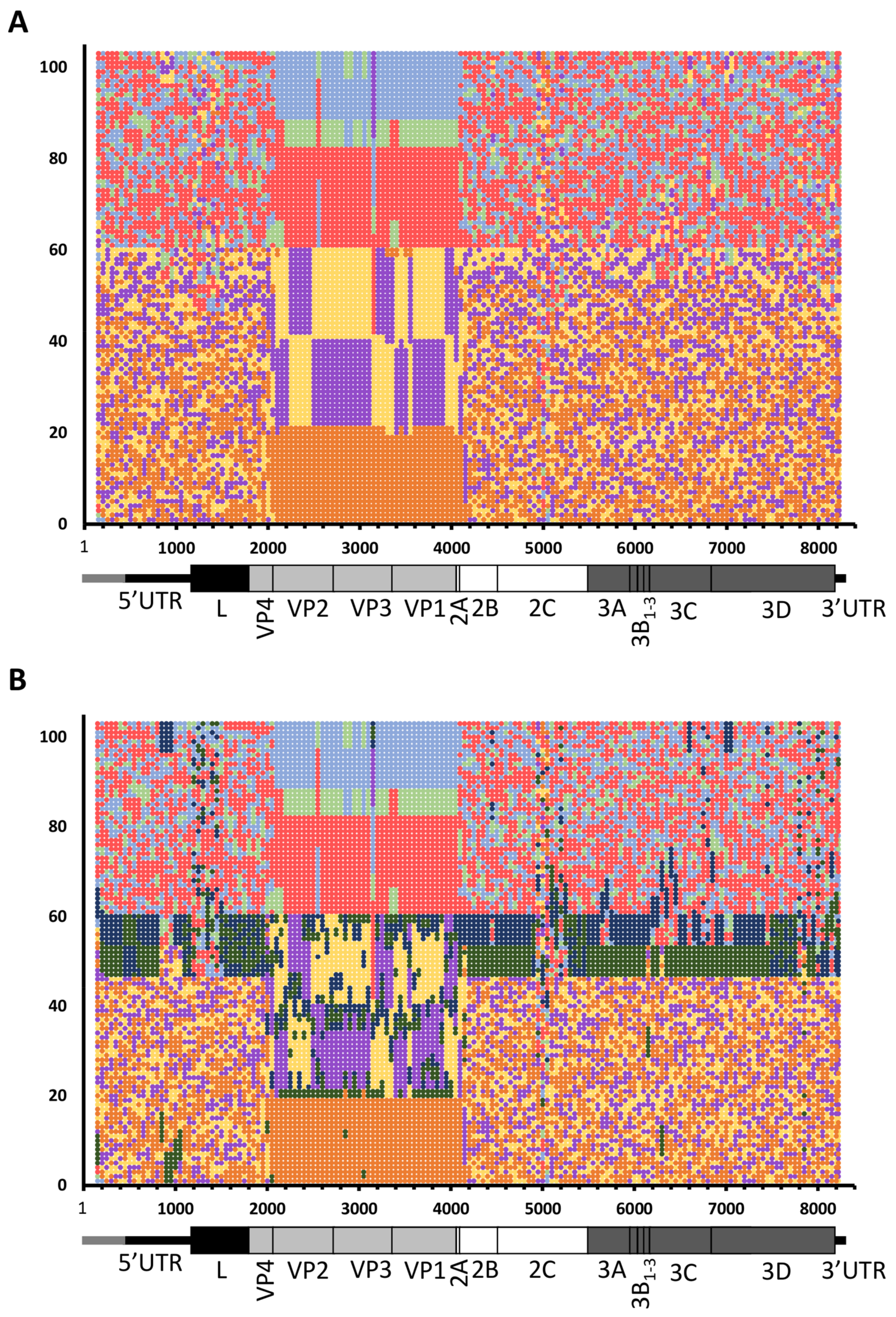


{kind=link}
{kind=link}
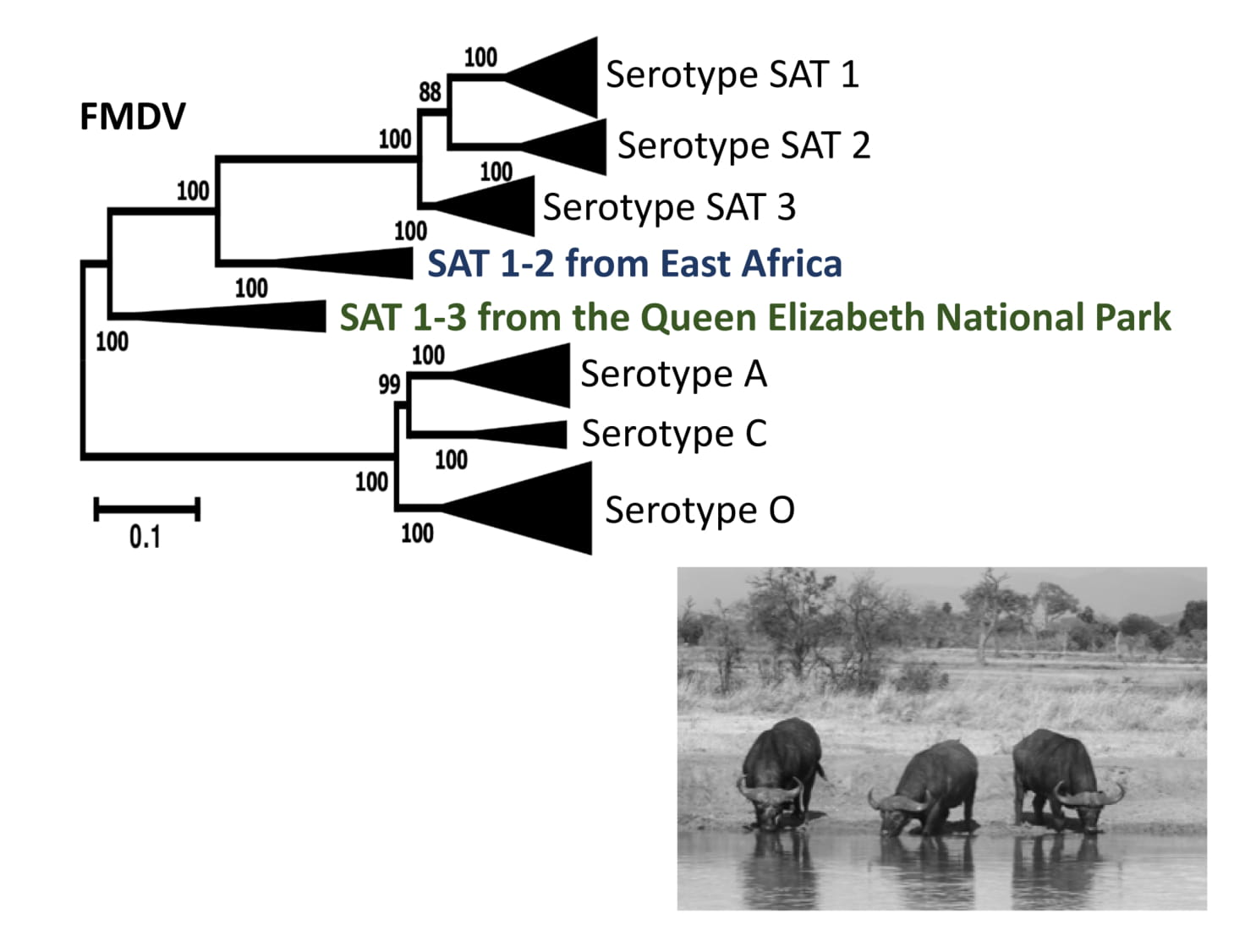
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serotype | Isolate Name | Year of Sampling | Country of Origin | Topotype | Host Species | GenBank Accession No. |
|---|---|---|---|---|---|---|
| A | EGY/1/72 | 1972 | Egypt | AFRICA | cattle | MH053305 |
| A | TCH/2/73 | 1973 | Chad | AFRICA | cattle | MH053306 |
| A | Zambia/90 | 1990 | Zambia | AFRICA | cattle | MH053307 |
| C | ETH/1/71 | 1971 | Ethiopia | AFRICA | not known | MH053308 |
| C | KEN/32/70 (K267/67) | 1967 | Kenya | AFRICA | cattle | MH053309 |
| C | UGA/18/70 | 1970 | Uganda | AFRICA | cattle | MH053310 |
| O | ETH/3/2004 | 2004 | Ethiopia | EA-3 | cattle | MH053311 |
| O | ETH/58/2005 | 2005 | Ethiopia | EA-4 | cattle | MH053312 |
| O | ETH/2/2006 | 2006 | Ethiopia | EA-3 | cattle | MH053313 |
| O | ETH/1/2007 | 2007 | Ethiopia | EA-3 | cattle | MH053314 |
| O | SUD/1/76 | 1976 | Sudan | WA | not known | MH053315 |
| O | UGA/5/96 | 1996 | Uganda | EA-1 | cattle | MH053316 |
| O | UGA/17/98 | 1998 | Uganda | EA-4 | not known | MH053317 |
| O | UGA/3/2002 | 2002 | Uganda | EA-2 | not known | MH053318 |
| SAT 1 | BOT-BUFF/62/74 | 1974 | Botswana | III (WZ) | Syncerus caffer | MH053319 |
| SAT 1 | K303/83 | 1983 | Kenya | I (NWZ) | cattle | MH053320 |
| SAT 1 | MOZ/5/81 | 1981 | Mozambique | II (SEZ) | cattle | MH053321 |
| SAT 1 | SWA/2/89 | 1989 | Nambia | III (WZ) | Syncerus caffer | MH053322 |
| SAT 1 | TCH/1/72 | 1972 | Chad | XI | cattle | MH053323 |
| SAT 1 | UGA/47/71 | 1971 | Uganda | VII (EA-2) | cattle | MH053324 |
| SAT 1 | UGA/6/78 | 1978 | Uganda | VII (EA-2) | cattle | MH053325 |
| SAT 1 | UGA-BUFF/10/70 | 1970 | Uganda | IV (EA-1) | Syncerus caffer | MH053326 |
| SAT 1 | UGA-BUFF/21/70 | 1970 | Uganda | IV (EA-1) | Syncerus caffer | MH053327 |
| SAT 2 | BOT-BUFF/2/68 | 1968 | Botswana | III | Syncerus caffer | MH053328 |
| SAT 2 | BOT-BUFF/2/69 | 1969 | Botswana | III | Syncerus caffer | MH053329 |
| SAT 2 | BOT-BUFF/17/69 | 1969 | Botswana | II | Syncerus caffer | MH053330 |
| SAT 2 | BOT-BUFF/107/72 | 1972 | Botswana | II | Syncerus caffer | MH053331 |
| SAT 2 | BOT-BUFF/170/74 | 1974 | Botswana | II | Syncerus caffer | MH053332 |
| SAT 2 | ETH/1/90 | 1989 | Ethiopia | IV | cattle | MH053333 |
| SAT 2 | NR/1/64 | 1964 | Zambia (Northern Rhodesia) | III | cattle | MH053334 |
| SAT 2 | SRHO/1/65 | 1965 | Zimbabwe (Southern Rhodesia) | I | cattle | MH053335 |
| SAT 2 | UGA-BUFF/12/70 | 1970 | Uganda | X | Syncerus caffer | MH053336 |
| SAT 2 | UGA-BUFF/24/70 | 1970 | Uganda | X | Syncerus caffer | MH053337 |
| SAT 3 | BOT/109/66 | 1966 | Botswana | II (WZ) | cattle | MH053338 |
| SAT 3 | BOT-BUFF/13/70 | 1970 | Botswana | II (WZ) | Syncerus caffer | MH053339 |
| SAT 3 | MAL/3/76 | 1976 | Malawi | III (NWZ) | cattle | MH053340 |
| SAT 3 | UGA-BUFF/27/70 | 1970 | Uganda | V | Syncerus caffer | MH053341 |
| SAT 3 | ZAM/P2/96 (NAN-11) | 1996 | Zambia | IV | Syncerus caffer | MH053342 |
| SAT 3 | RV/7/34 | 1934 | Zimbabwe (Rhodesia) | VI | cattle | MH053343 |
| SAT 3 | RHO/7/74 | 1974 | Zimbabwe (Rhodesia) | I (SEZ) | cattle | MH053344 |
| SAT 3 | RHO/5/75 | 1975 | Zimbabwe (Rhodesia) | I (SEZ) | cattle | MH053345 |
| SAT 3 | RHO/16/76 | 1976 | Zimbabwe (Rhodesia) | I (SEZ) | cattle | MH053346 |
| SAT 3 | RHO/23/76 | 1976 | Zimbabwe (Rhodesia) | I (SEZ) | cattle | MH053347 |
| SAT 3 | RHO/11/77 | 1977 | Zimbabwe (Rhodesia) | I (SEZ) | cattle | MH053348 |
| SAT 3 | ZIM/P6/83 (WN-16) | 1983 | Zimbabwe | I (SEZ) | Syncerus caffer | MH053349 |
| SAT 3 | ZIM/P6/83 (WN-19) | 1983 | Zimbabwe | I (SEZ) | Syncerus caffer | MH053350 |
| SAT 3 | ZIM/2/84 | 1984 | Zimbabwe | II (WZ) | cattle | MH053351 |
| SAT 3 | ZIM/P27/90 (DSA-31) | 1990 | Zimbabwe | III (NWZ) | Syncerus caffer | MH053352 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lasecka-Dykes, L.; Wright, C.F.; Di Nardo, A.; Logan, G.; Mioulet, V.; Jackson, T.; Tuthill, T.J.; Knowles, N.J.; King, D.P. Full Genome Sequencing Reveals New Southern African Territories Genotypes Bringing Us Closer to Understanding True Variability of Foot-and-Mouth Disease Virus in Africa. Viruses 2018, 10, 192. https://doi.org/10.3390/v10040192
Lasecka-Dykes L, Wright CF, Di Nardo A, Logan G, Mioulet V, Jackson T, Tuthill TJ, Knowles NJ, King DP. Full Genome Sequencing Reveals New Southern African Territories Genotypes Bringing Us Closer to Understanding True Variability of Foot-and-Mouth Disease Virus in Africa. Viruses. 2018; 10(4):192. https://doi.org/10.3390/v10040192
Chicago/Turabian StyleLasecka-Dykes, Lidia, Caroline F. Wright, Antonello Di Nardo, Grace Logan, Valerie Mioulet, Terry Jackson, Tobias J. Tuthill, Nick J. Knowles, and Donald P. King. 2018. "Full Genome Sequencing Reveals New Southern African Territories Genotypes Bringing Us Closer to Understanding True Variability of Foot-and-Mouth Disease Virus in Africa" Viruses 10, no. 4: 192. https://doi.org/10.3390/v10040192
APA StyleLasecka-Dykes, L., Wright, C. F., Di Nardo, A., Logan, G., Mioulet, V., Jackson, T., Tuthill, T. J., Knowles, N. J., & King, D. P. (2018). Full Genome Sequencing Reveals New Southern African Territories Genotypes Bringing Us Closer to Understanding True Variability of Foot-and-Mouth Disease Virus in Africa. Viruses, 10(4), 192. https://doi.org/10.3390/v10040192