Single Viruses on the Fluorescence Microscope: Imaging Molecular Mobility, Interactions and Structure Sheds New Light on Viral Replication


Abstract
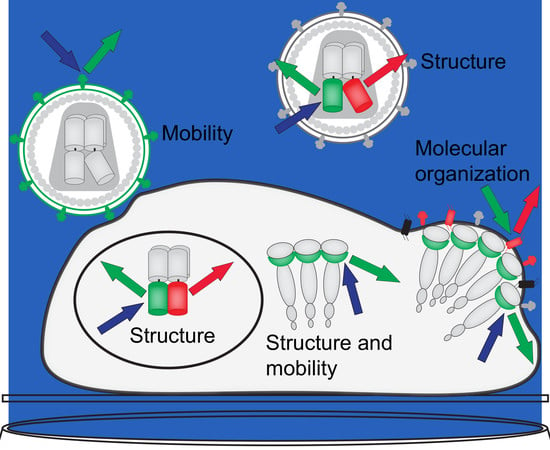
1. Introduction
2. Single Virus Imaging Hardware
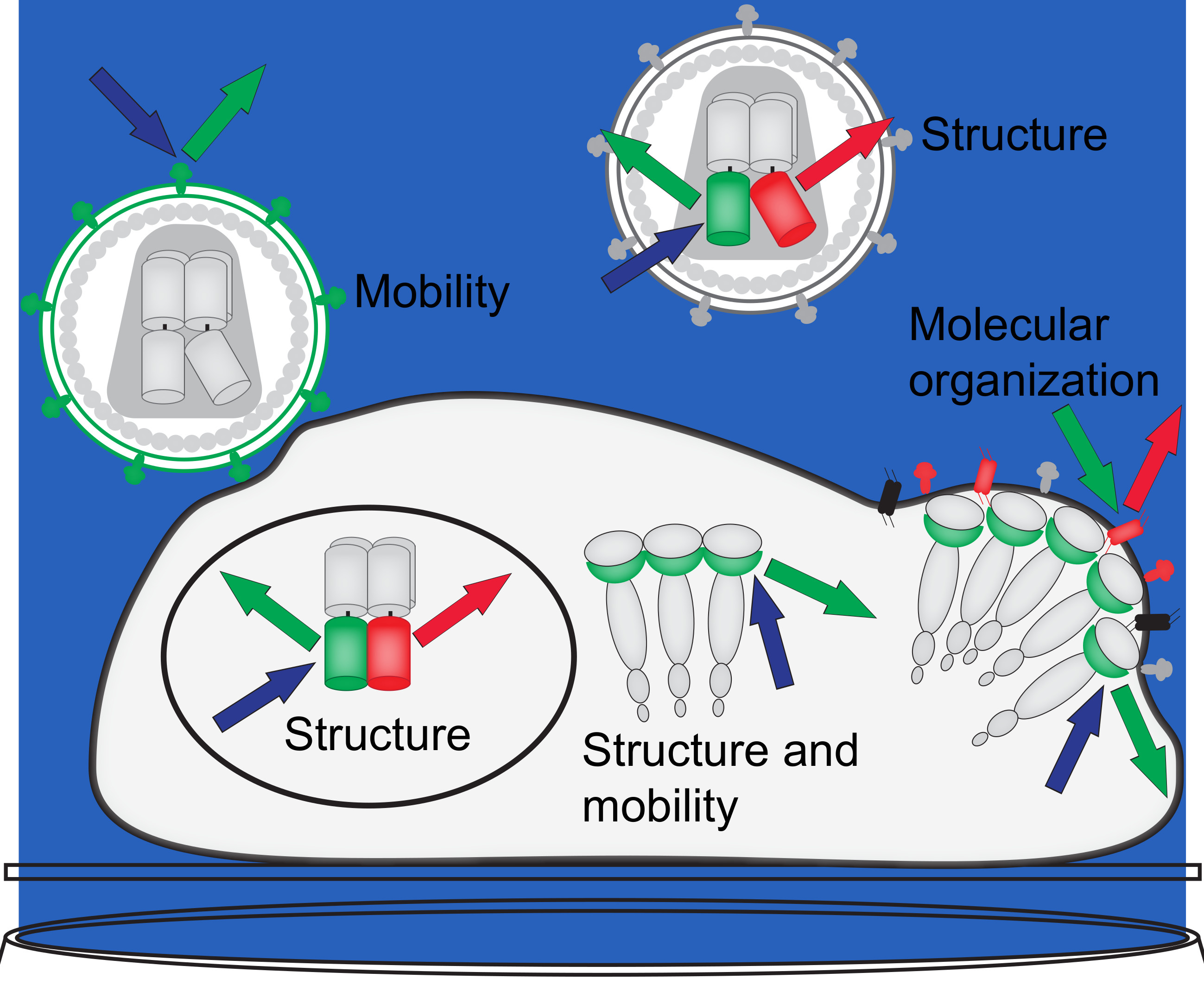
3. Förster Resonance Energy Transfer Probes the Human Immunodeficiency Virus (HIV-1) Integrase Quaternary Structure
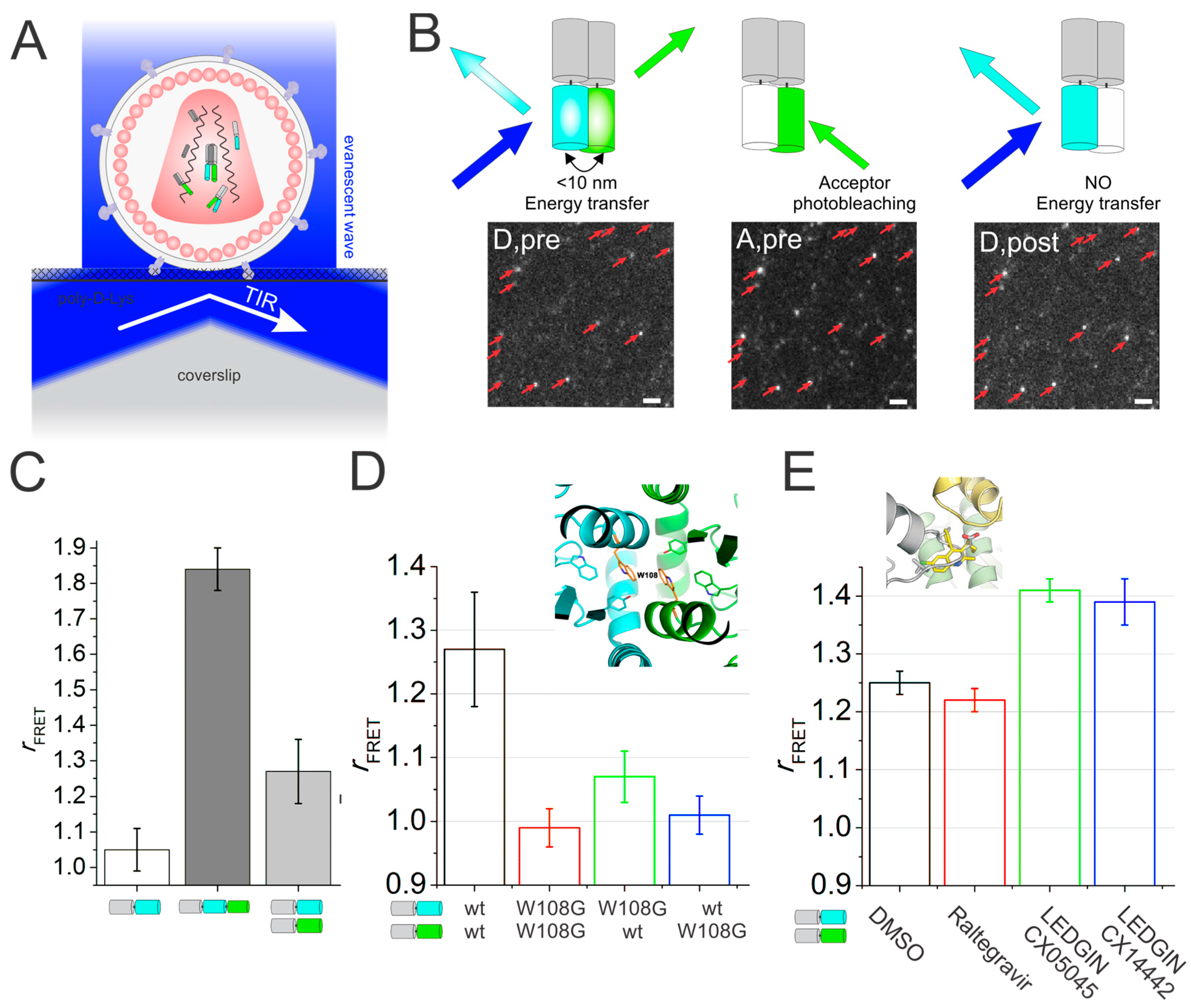
4. Single Particle Tracking to Study Simian Virus 40 Membrane Attachment
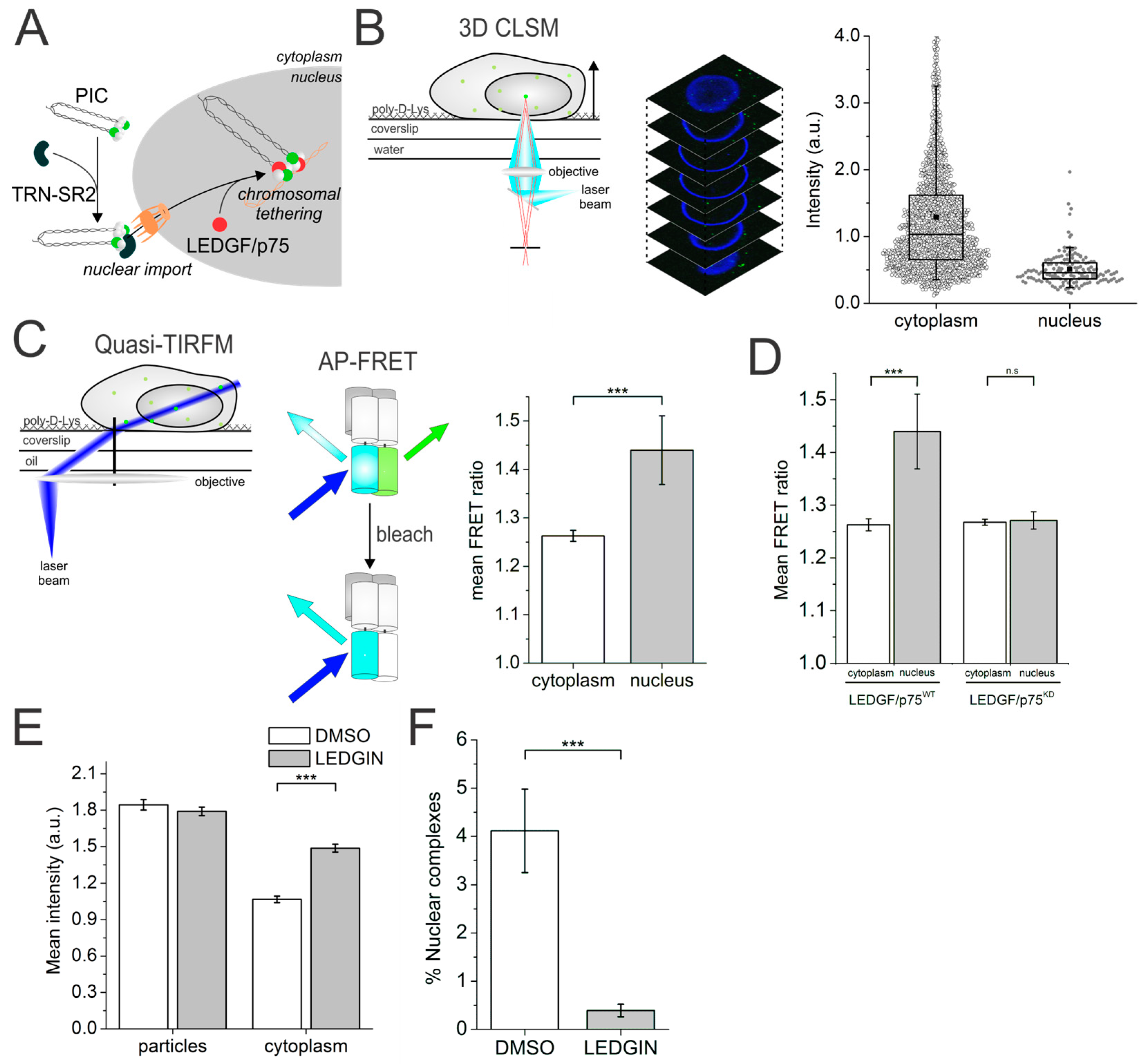
5. Quantitative Confocal Microscopy and Förster Resonance Energy Transfer (FRET) Reveals Dynamic Integrase (IN) Oligomerization
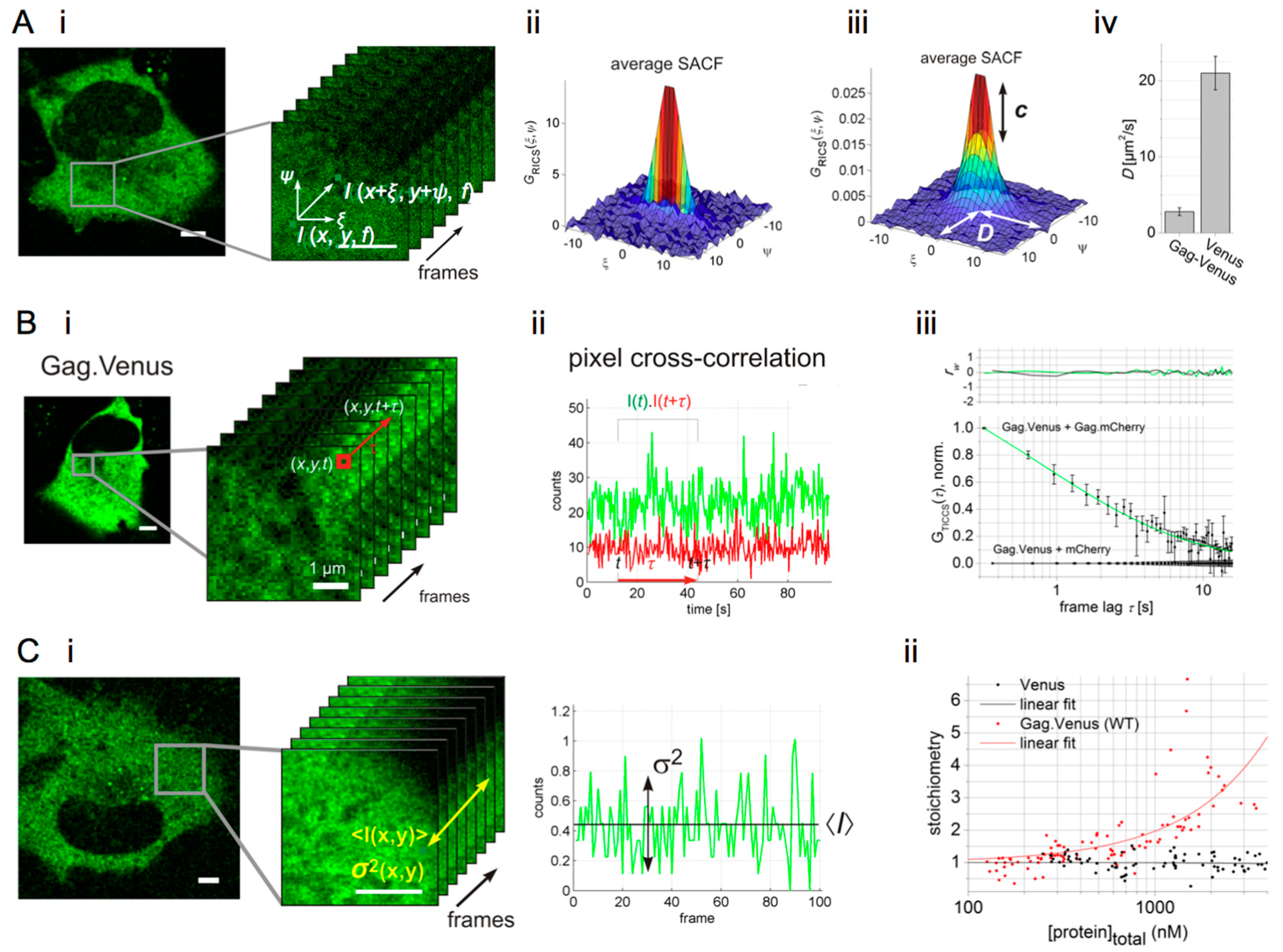
6. Image Correlation Spectroscopy Reveals Cytosolic Assembly of HIV-1 Gag
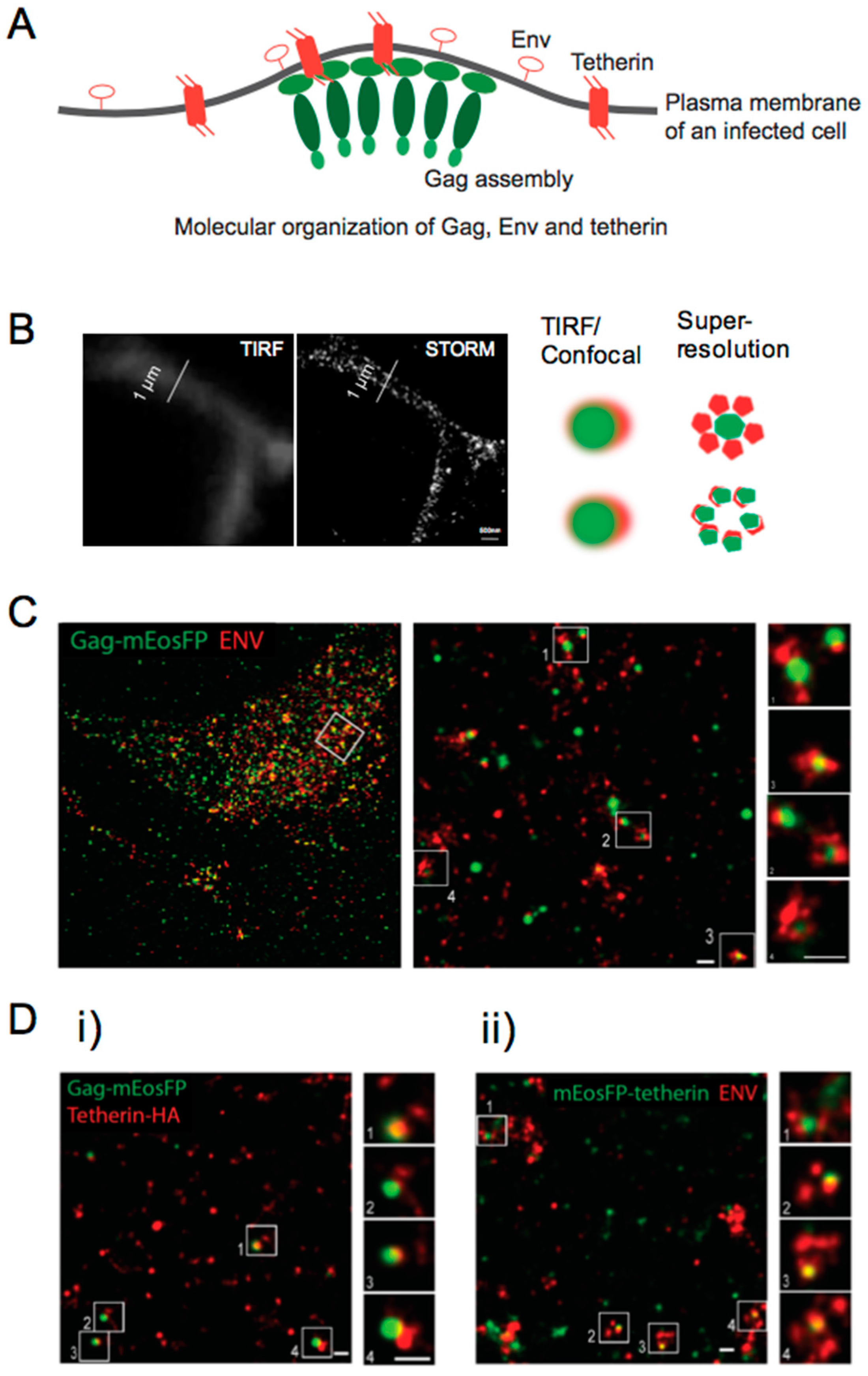
7. Super-Resolution Microscopy Sheds Light on Viral Restriction at the Plasma Membrane
8. General Conclusions and Outlook
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Cui, J.; Schlub, T.E.; Holmes, E.C. An allometric relationship between the genome length and virion volume of viruses. J. Virol. 2014, 88, 6403–6410. [Google Scholar] [CrossRef] [PubMed]
- Klasse, P.J. Molecular determinants of the ratio of inert to infectious virus particles. In Molecular Basis of Viral Infection; Klasse, P.J., Ed.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2015; Volume 129, pp. 285–326. [Google Scholar] [CrossRef]
- Flint, S.J.; Enquist, L.W.; Racaniello, V.R.; Skalka, A.M. Principles of virology. In American Society for Microbiology, 3rd ed.; Academic Press: ASM press, Washington, DC, USA, 2009. [Google Scholar]
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-S.; Hughes, S.H. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.; Maghelli, N.; Tolic-Norrelykke, I.M. Single-molecule imaging in vivo: The dancing building blocks of the cell. Integr. Biol. 2013, 5, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Hanne, J.; Zila, V.; Heilemann, M.; Muller, B.; Krausslich, H.G. Super-resolved insights into human immunodeficiency virus biology. FEBS Lett. 2016, 590, 1858–1876. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.F.; Rossy, J.; Owen, D.M.; Mak, J.; Gaus, K. HIV taken by storm: Super-resolution fluorescence microscopy of a viral infection. Virol. J. 2012, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Sauer, M.; Heilemann, M. Single-molecule localization microscopy in eukaryotes. Chem. Rev. 2017, 117, 7478–7509. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Dalal, R.; Horwitz, A.F.; Gratton, E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008, 94, 2320–2332. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Gratton, E. Fluorescence correlation spectroscopy and fluorescence cross-correlation spectroscopy. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Wiseman, P.W.; Choi, C.; Horwitz, A.R.; Gratton, E. Stoichiometry of molecular complexes at adhesions in living cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2170–2175. [Google Scholar] [CrossRef] [PubMed]
- Kenworthy, A.K. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods 2001, 24, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.R.; Ma, Y.M.; Vogt, V.M.; Webb, W.W. Direct measurement of gag-gag interaction during retrovirus assembly with fret and fluorescence correlation spectroscopy. J. Cell Biol. 2003, 162, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Nath, A.; Farber, M.; Datta, S.A.K.; Rein, A.; Rhoades, E.; Mothes, W. A conformational transition observed in single HIV-1 gag molecules during in vitro assembly of virus-like particles. J. Virol. 2014, 88, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Borrenberghs, D.; Dirix, L.; De Wit, F.; Rocha, S.; Blokken, J.; De Houwer, S.; Gijsbers, R.; Christ, F.; Hofkens, J.; Hendrix, J.; et al. Dynamic oligomerization of integrase orchestrates HIV nuclear entry. Sci. Rep. 2016, 6, 36485. [Google Scholar] [CrossRef] [PubMed]
- Borrenberghs, D.; Thys, W.; Rocha, S.; Demeulemeester, J.; Weydert, C.; Dedecker, P.; Hofkens, J.; Debyser, Z.; Hendrix, J. HIV virions as nanoscopic test tubes for probing oligomerization of the integrase enzyme. ACS Nano 2014, 8, 3531–3545. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, J.; Baumgartel, V.; Schrimpf, W.; Ivanchenko, S.; Digman, M.A.; Gratton, E.; Krausslich, H.G.; Muller, B.; Lamb, D.C. Live-cell observation of cytosolic HIV-1 assembly onset reveals rna-interacting gag oligomers. J. Cell Biol. 2015, 210, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Rocha, S.; Mangeat, B.; Blanchet, F.; Uji-i, H.; Hofkens, J.; Piguet, V. Quantitative multicolor super-resolution microscopy reveals tetherin HIV-1 interaction. PLoS Pathog. 2011, 7, e1002456. [Google Scholar] [CrossRef] [PubMed]
- Parveen, N.; Block, S.; Zhdanov, V.P.; Rydel, G.E.; Hook, F. Detachment of membrane bound virions by competitive ligand binding induced receptor depletion. Langmuir 2017, 33, 4049–4056. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.O. Integration. In Retriviruses; Coffin, J., Hughes, S., Varmus, H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Craigie, R. HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. 2001, 276, 23213–23216. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.; Schwartz, O.; Mammano, F. Oligomerization within virions and subcellular localization of human immunodeficiency virus type 1 integrase. J. Virol. 1999, 73, 5079–5088. [Google Scholar] [PubMed]
- Chen, J.C.H.; Krucinski, J.; Miercke, L.J.W.; Finer-Moore, J.S.; Tang, A.H.; Leavitt, A.D.; Stroud, R.M. Crystal structure of the HIV-1 integrase catalytic core and c-terminal domains: A model for viral DNA binding. Proc. Natl. Acad. Sci. USA 2000, 97, 8233–8238. [Google Scholar] [CrossRef] [PubMed]
- Deprez, E.; Tauc, P.; Leh, H.; Mouscadet, J.F.; Auclair, C.; Brochon, J.C. Oligomeric states of the HIV-1 integrase as measured by time-resolved fluorescence anisotropy. Biochemistry 2000, 39, 9275–9284. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Bushman, F.D.; Craigie, R. Identification of discrete functional domains of HIV-1 integrase and their organization within an active multimeric complex. EMBO J. 1993, 12, 3269–3275. [Google Scholar] [PubMed]
- Guiot, E.; Carayon, K.; Delelis, O.; Simon, F.; Tauc, P.; Zubin, E.; Gottikh, M.; Mouscadet, J.F.; Brochon, J.C.; Deprez, E. Relationship between the oligomeric status of HIV-1 integrase on DNA and enzymatic activity. J. Biol. Chem. 2006, 281, 22707–22719. [Google Scholar] [CrossRef] [PubMed]
- Goldgur, Y.; Craigie, R.; Cohen, G.H.; Fujiwara, T.; Yoshinaga, T.; Fujishita, T.; Sugimoto, H.; Endo, T.; Murai, H.; Davies, D.R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: A platform for antiviral drug design. Proc. Natl. Acad. Sci. USA 1999, 96, 13040–13043. [Google Scholar] [CrossRef] [PubMed]
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.F.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Ling, H.; Yang, W.; Craigie, R. Structure of a two-domain fragment of HIV-1 integrase: Implications for domain organization in the intact protein. EMBO J. 2001, 20, 7333–7343. [Google Scholar] [CrossRef] [PubMed]
- Sisamakis, E.; Valeri, A.; Kalinin, S.; Rothwell, P.J.; Seidel, C.A.M. Accurate single-molecule fret studies using multiparameter fluorescence detection. In Methods in Enzymology, Vol 475: Single Molecule Tools, pt b: Super-Resolution, Particle Tracking, Multiparameter, and Force Based Methods; Walter, N.G., Ed.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2010; Volume 475, pp. 455–514. [Google Scholar] [CrossRef]
- Herschhorn, A.; Ma, X.; Gu, C.; Ventura, J.D.; Castillo-Menendez, L.; Melillo, B.; Terry, D.S.; Smith, A.B.; Blanchard, S.C.; Munro, J.B. Release of gp120 restraints leads to an entry-competent intermediate state of the HIV-1 envelope glycoproteins. MBio 2016, 7, e01598-01516. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.M.; Padilla-Parra, S. Imaging real-time HIV-1 virion fusion with fret-based biosensors. Sci. Rep. 2015, 5, 13449. [Google Scholar] [CrossRef] [PubMed]
- Sood, C.; Francis, A.C.; Desai, T.M.; Melikyan, G.B. An improved labeling strategy enables automated detection of single-virus fusion and assessment of HIV-1 protease activity in single virions. J. Biol. Chem. 2017, 292, 20196–20207. [Google Scholar] [CrossRef] [PubMed]
- Abbondanzieri, E.A.; Bokinsky, G.; Rausch, J.W.; Zhang, J.X.; Le Grice, S.F.J.; Zhuang, X.W. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature 2008, 453, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Abbondanzieri, E.A.; Rausch, J.W.; Le Grice, S.F.; Zhuang, X. Slide into action: Dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science 2008, 322, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Day, R.N.; Booker, C.F.; Periasamy, A. Characterization of an improved donor fluorescent protein for förster resonance energy transfer microscopy. J. Biomed. Opt. 2008, 13, 031203. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Albanese, A.; Arosio, D.; Terreni, M.; Cereseto, A. HIV-1 pre-integration complexes selectively target decondensed chromatin in the nuclear periphery. PLoS ONE 2008, 3, e2413. [Google Scholar] [CrossRef] [PubMed]
- Serrao, E.; Thys, W.; Demeulemeester, J.; Al-Mawsawi, L.Q.; Christ, F.; Debyser, Z.; Neamati, N. A symmetric region of the HIV-1 integrase dimerization interface is essential for viral replication. PLoS ONE 2012, 7, e45177. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Desimmie, B.A.; Schrijvers, R.; Demeulemeester, J.; Borrenberghs, D.; Weydert, C.; Thys, W.; Vets, S.; Van Remoortel, B.; Hofkens, J.; De Rijck, J.; et al. Ledgins inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 2013, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Jena, N.; Koh, Y.; Taskent-Sezgin, H.; Slaughter, A.; Feng, L.; de Silva, S.; Wu, L.; Le Grice, S.F.J.; Engelman, A.; et al. Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. [Google Scholar] [CrossRef] [PubMed]
- Tsiang, M.; Jones, G.S.; Niedziela-Majka, A.; Kan, E.; Lansdon, E.B.; Huang, W.N.; Hung, M.; Samuel, D.; Novikov, N.; Xu, Y.L.; et al. New class of HIV-1 integrase (in) inhibitors with a dual mode of action. J. Biol. Chem. 2012, 287, 21189–21203. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Marsh, M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Stanifer, M.; Lozach, P.-Y. Dynamics of virus-receptor interactions in virus binding, signaling, and endocytosis. Viruses 2015, 7, 2794–2815. [Google Scholar] [CrossRef] [PubMed]
- Sieben, C.; Kappel, C.; Zhu, R.; Wozniak, A.; Rankl, C.; Hinterdorfer, P.; Grubmüller, H.; Herrmann, A. Influenza virus binds its host cell using multiple dynamic interactions. Proc. Natl. Acad. Sci. 2012, 109, 13626. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, O.M.; Gonzalez-Segredo, N.; Kukura, P.; Oppenheim, A.; Choquet, D.; Sandoghdar, V.; Helenius, A.; Sbalzarini, I.F.; Ewers, H. Receptor concentration and diffusivity control multivalent binding of sv40 to membrane bilayers. PLoS Comput. Biol. 2013, 9, e1003310. [Google Scholar] [CrossRef] [PubMed]
- Ewers, H.; Smith, A.E.; Sbalzarini, I.F.; Lilie, H.; Koumoutsakos, P.; Helenius, A. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc. Natl. Acad. Sci. USA 2005, 102, 15110–15115. [Google Scholar] [CrossRef] [PubMed]
- Cheezum, M.K.; Walker, W.F.; Guilford, W.H. Quantitative comparison of algorithms for tracking single fluorescent particles. Biophys. J. 2001, 81, 2378–2388. [Google Scholar] [CrossRef]
- Chenouard, N.; Smal, I.; de Chaumont, F.; Maska, M.; Sbalzarini, I.F.; Gong, Y.H.; Cardinale, J.; Carthel, C.; Coraluppi, S.; Winter, M.; et al. Objective comparison of particle tracking methods. Nat. Methods 2014, 11, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, A.; Selvin, P.R. Fluorescence imaging with one nanometer accuracy: Application to molecular motors. Acc. Chem. Res. 2005, 38, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, B.; Zhuang, X.W. Virus trafficking—Learning from single-virus tracking. Nat. Rev. Microbiol. 2007, 5, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Ruthardt, N.; Lamb, D.C.; Brauchle, C. Single-particle tracking as a quantitative microscopy-based approach to unravel cell entry mechanisms of viruses and pharmaceutical nanoparticles. Mol. Ther. 2011, 19, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Saxton, M.J.; Jacobson, K. Single-particle tracking:Applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X.W. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar] [CrossRef] [PubMed]
- Peerboom, N.; Block, S.; Altgarde, N.; Wahlsten, O.; Moller, S.; Schnabelrauch, M.; Trybala, E.; Bergstrom, T.; Bally, M. Binding kinetics and lateral mobility of hsv-1 on end-grafted sulfated glycosaminoglycans. Biophys. J. 2017, 113, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N.; Genovesio, A.; Kim, K.A.; Miko, S.; Perret, E.; Olivo-Marin, J.C.; Shorte, S.; Charneau, P. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 2006, 3, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, G.; Ried, M.U.; Endreß, T.; Büning, H.; Hallek, M.; Bräuchle, C. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science 2001, 294, 1929. [Google Scholar] [CrossRef] [PubMed]
- Arcizet, D.; Meier, B.; Sackmann, E.; Radler, J.O.; Heinrich, D. Temporal analysis of active and passive transport in living cells. Phys. Rev. Lett. 2008, 101, 248103. [Google Scholar] [CrossRef] [PubMed]
- Block, S.; Zhdanov, V.P.; Höök, F. Quantification of multivalent interactions by tracking single biological nanoparticle mobility on a lipid membrane. Nano Lett. 2016. [Google Scholar] [CrossRef] [PubMed]
- Saxton, M.J. Single-particle tracking: The distribution of diffusion coefficients. Biophys. J. 1997, 72, 1744–1753. [Google Scholar] [CrossRef]
- Francis, A.C.; Marin, M.; Shi, J.; Aiken, C.; Melikyan, G.B. Time-resolved imaging of single HIV-1 uncoating in vitro and in living cells. PLoS Pathog. 2016, 12, e1005709. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, C.J.; Greber, U.F. Virus movements on the plasma membrane support infection and transmission between cells. PLoS Pathog. 2009, 5, e1000621. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N. Revisiting HIV-1 uncoating. Retrovirology 2010, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- De Rijck, J.; Vandekerckhove, L.; Christ, F.; Debyser, Z. Lentiviral nuclear import: A complex interplay between virus and host. Bioessays 2007, 29, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Hilditch, L.; Towers, G.J. A model for cofactor use during HIV-1 reverse transcription and nuclear entry. Curr. Opin. Virol. 2014, 4, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Ao, Z.J.; Jayappa, K.D.; Wang, B.C.; Zheng, Y.F.; Kung, S.; Rassart, E.; Depping, R.; Kohler, M.; Cohen, E.A.; Yao, X.J. Importin alpha 3 interacts with HIV-1 integrase and contributes to HIV-1 nuclear import and replication. J. Virol. 2010, 84, 8650–8663. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Thys, W.; De Rijck, J.; Gijsbers, R.; Albanese, A.; Arosio, D.; Emiliani, S.; Rain, J.C.; Benarous, R.; Cereseto, A.; et al. Transportin-sr2 imports HIV into the nucleus. Curr. Biol. 2008, 18, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A. The roles of cellular factors in retroviral integration. Curr.Top. Microbiol. Immunol. 2003, 281, 209–238. [Google Scholar] [PubMed]
- Taltynov, O.; Desimmie, B.A.; Demeulemeester, J.; Christ, F.; Debyser, Z. Cellular cofactors of lentiviral integrase: From target validation to drug discovery. Mol. Biol. Int. 2012, 2012, 863405. [Google Scholar] [CrossRef] [PubMed]
- Tsirkone, V.G.; Blokken, J.; De Wit, F.; Breemans, J.; De Houwer, S.; Debyser, Z.; Christ, F.; Strelkov, S.V. N-terminal half of transportin sr2 interacts with HIV integrase. J. Biol. Chem. 2017, 292, 9699–9710. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with ledgf/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the ledgf/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- McKee, C.J.; Kessl, J.J.; Shkriabai, N.; Dar, M.J.; Engelman, A.; Kvaratskhelia, M. Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (ledgf) protein. J. Biol. Chem. 2008, 283, 31802–31812. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Imamoto, N.; Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 2008, 5, 159. [Google Scholar] [CrossRef] [PubMed]
- Jurado, K.A.; Engelman, A. Multimodal mechanism of action of allosteric HIV-1 integrase inhibitors. Expert Rev. Mol. Med. 2013, 15, e14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baumgärtel, V.; Ivanchenko, S.; Dupont, A.; Sergeev, M.; Wiseman, P.W.; Kräusslich, H.-G.; Bräuchle, C.; Müller, B.; Lamb, D.C. Live-cell visualization of dynamics of HIV budding site interactions with an escrt component. Nat. Cell Biol. 2011, 13, 469. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Delviks-Frankenberry, K.A.; Chen, J.; Janaka, S.K.; Sastri, J.; Hu, W.-S.; Pathak, V.K. Dynamics and regulation of nuclear import and nuclear movements of HIV-1 complexes. PLoS Pathog. 2017, 13, e1006570. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 2009, 106, 19114–19119. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; He, Z.; Tan, T.; Li, W.; Zhang, Z.; Song, S.; Zhang, X.; Hu, Q.; Zhou, P.; Wu, Y.; et al. Real-time imaging of single HIV-1 disassembly with multicolor viral particles. ACS Nano 2016, 10, 6273–6282. [Google Scholar] [CrossRef] [PubMed]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef] [PubMed]
- Candotti, F.; Shaw, K.L.; Muul, L.; Carbonaro, D.; Sokolic, R.; Choi, C.; Schurman, S.H.; Garabedian, E.; Kesserwan, C.; Jagadeesh, G.J.; et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: Clinical comparison of retroviral vectors and treatment plans. Blood 2012, 120, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.A.; Lemischka, I.R.; Nathan, D.G.; Mulligan, R.C. Introduction of new genetic material into pluripotent hematopoietic stem-cells of the mouse. Nature 1984, 310, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.; Thrasher, A. Gene therapy: Progress and predictions. Proc. R. Soc. B-Biol. Sci. 2015, 282, 8. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. Lmo2-associated clonal t cell proliferation in two patients after gene therapy for scid-x1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of scid-x1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Borrenberghs, D.; Dirix, L.; Cereseto, A.; Hofkens, J.; Debyser, Z.; Hendrix, J. Post-mitotic bet-induced reshaping of integrase quaternary structure supports wild-type MLV integration. 2018; Submitted. [Google Scholar]
- Bell, N.M.; Lever, A.M.L. HIV gag polyprotein: Processing and early viral particle assembly. Trends Microbiol. 2013, 21, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Baumgartel, V.; Muller, B.; Lamb, D.C. Quantitative live-cell imaging of human immunodeficiency virus (HIV-1) assembly. Viruses 2012, 4, 777–799. [Google Scholar] [CrossRef] [PubMed]
- Ganser-Pornillos, B.K.; Yeager, M.; Pornillos, O. Assembly and architecture of HIV. In Viral Molecular Machines; Rossmann, M.G., Rao, V.B., Eds.; Springer-Verlag: Berlin, Germany, 2012; Volume 726, pp. 441–465. [Google Scholar] [CrossRef]
- Muriaux, D.; Darlix, J.L. Properties and functions of the nucleocapsid protein in virus assembly. RNA Biol. 2010, 7, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Rein, A.; Datta, S.A.K.; Jones, C.P.; Musier-Forsyth, K. Diverse interactions of retroviral gag proteins with RNAS. Trends Biochem. Sci. 2011, 36, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Ivanchenko, S.; Godinez, W.J.; Lampe, M.; Kräusslich, H.-G.; Eils, R.; Rohr, K.; Bräuchle, C.; Müller, B.; Lamb, D.C. Dynamics of HIV-1 assembly and release. PLoS Pathog. 2009, 5, e1000652. [Google Scholar] [CrossRef] [PubMed]
- Spearman, P. HIV-1 gag as an antiviral target: Development of assembly and maturation inhibitors. Curr. Top. Med. Chem. 2016, 16, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.A.; Freed, E.O. HIV type 1 gag as a target for antiviral therapy. AIDS Res. Hum. Retrovir. 2012, 28, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Ono, A. HIV-1 assembly at the plasma membrane: Gag trafficking and localization. Future Virol. 2009, 4, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef] [PubMed]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural basis for targeting HIV-1 gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Liu, B.D.; Yu, X.F. Formation of virus assembly intermediate complexes in the cytoplasm by wild-type and assembly-defective mutant human immunodeficiency virus type 1 and their association with membranes. J. Virol. 1999, 73, 5654–5662. [Google Scholar] [PubMed]
- Nermut, M.V.; Zhang, W.H.; Francis, G.; Ciampor, F.; Morikawa, Y.; Jones, I.M. Time course of gag protein assembly in HIV-1-infected cells: A study by immunoelectron microscopy. Virology 2003, 305, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, K.H.; Berk, S.; Grigsby, I.F.; Chen, Y.; Mansky, L.M.; Mueller, J.D. Interrelationship between cytoplasmic retroviral gag concentration and gag-membrane association. J. Mol. Biol. 2014, 426, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Dunne, P.D.; Fernandes, R.A.; McColl, J.; Yoon, J.W.; James, J.R.; Davis, S.J.; Klenerman, D. Dysco: Quantitating associations of membrane proteins using two-color single-molecule tracking. Biophys. J. 2009, 97, L5–L7. [Google Scholar] [CrossRef] [PubMed]
- Dupont, A.; Stirnnagel, K.; Lindemann, D.; Lamb, D.C. Tracking image correlation: Combining single-particle tracking and image correlation. Biophys. J. 2013, 104, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, D.; Deschout, H.; Remaut, K.; Engbersen, J.F.J.; Jones, A.T.; Demeester, J.; De Smedt, S.C.; Braeckmans, K. Dynamic colocalization microscopy to characterize intracellular trafficking of nanomedicines. ACS Nano 2011, 5, 7874–7884. [Google Scholar] [CrossRef] [PubMed]
- Haustein, E.; Schwille, P. Fluorescence correlation spectroscopy: Novel variations of an established technique. In Annual Review of Biophysics and Biomolecular Structure; Annual Reviews: Palo Alto, CA, USA, 2007; Volume 36, pp. 151–169. [Google Scholar] [CrossRef]
- Digman, M.A.; Stakic, M.; Gratton, E. Chapter six-raster image correlation spectroscopy and number and brightness analysis. In Methods in Enzymology; Sergey, Y.T., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 518, pp. 121–144. [Google Scholar] [CrossRef]
- Kim, S.A.; Heinze, K.G.; Schwille, P. Fluorescence correlation spectroscopy in living cells. Nat. Methods 2007, 4, 963. [Google Scholar] [CrossRef] [PubMed]
- Tetin, S.Y. Preface. In Methods in Enzymology; Sergey, Y.T., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 518, pp. xi–xii. [Google Scholar] [CrossRef]
- Bacia, K.; Kim, S.A.; Schwille, P. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods 2006, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Hebert, B.; Costantino, S.; Wiseman, P.W. Spatiotemporal image correlation spectroscopy (stics) theory, verification, and application to protein velocity mapping in living cho cells. Biophys. J. 2005, 88, 3601–3614. [Google Scholar] [CrossRef] [PubMed]
- Oehlenschläger, F.; Schwille, P.; Eigen, M. Detection of HIV-1 rna by nucleic acid sequence-based amplification combined with fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 12811–12816. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, B.; Musier-Forsyth, K.; Mansky, L.M.; Mueller, J.D. Fluorescence fluctuation spectroscopy on viral-like particles reveals variable gag stoichiometry. Biophys. J. 2009, 96, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, K.H.; Zhang, W.; Grigsby, I.F.; Johnson, J.L.; Chen, Y.; Mueller, J.D.; Mansky, L.M. New insights into htlv-1 particle structure, assembly, and gag-gag interactions in living cells. Viruses 2011, 3, 770–793. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, J.; Gijsbers, R.; De Rijck, J.; Voet, A.; Hotta, J.I.; McNeely, M.; Hofkens, J.; Debyser, Z.; Engelborghs, Y. The transcriptional co-activator ledgf/p75 displays a dynamic scan-and-lock mechanism for chromatin tethering. Nucleic Acids Res. 2011, 39, 1310–1325. [Google Scholar] [CrossRef] [PubMed]
- Krieger, J.W.; Singh, A.P.; Bag, N.; Garbe, C.S.; Saunders, T.E.; Langowski, J.; Wohland, T. Imaging fluorescence (cross-) correlation spectroscopy in live cells and organisms. Nat. Protoc. 2015, 10, 1948. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Petersen, N.O. Diffusion of transferrin receptor clusters. Biophys. Chem. 1998, 75, 201–211. [Google Scholar] [CrossRef]
- Wiseman, P.W.; Squier, J.A.; Ellisman, M.H.; Wilson, K.R. Two-photon image correlation spectroscopy and image cross-correlation spectroscopy. J. Microsc.-Oxf. 2000, 200, 14–25. [Google Scholar] [CrossRef]
- Hendrix, J.; Schrimpf, W.; Holler, M.; Lamb, D.C. Pulsed interleaved excitation fluctuation imaging. Biophys. J. 2013, 105, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Grove, J. Super-resolution microscopy: A virus’ eye view of the cell. Viruses 2014, 6, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Vangindertael, J.; Camacho, R.; Sempels, W.; Mizuno, H.; Dedecker, P.; Janssen, K.P.F. An introduction to optical super-resolution microscopy for the adventurous biologist. Methods Appl. Fluoresc. 2018. [Google Scholar] [CrossRef] [PubMed]
- McKinney, S.A.; Murphy, C.S.; Hazelwood, K.L.; Davidson, M.W.; Looger, L.L. A bright and photostable photoconvertible fluorescent protein. Nat. Methods 2009, 6, 131. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; De Keersmaecker, H.; Uji-i, H.; Hofkens, J.; Mizuno, H. Photoswitchable fluorescent proteins for superresolution fluorescence microscopy circumventing the diffraction limit of light. In Fluorescence Spectroscopy and Microscopy: Methods and Protocols; Engelborghs, Y., Visser, A., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2014; Volume 1076, pp. 793–812. [Google Scholar] [CrossRef]
- Shcherbakova, D.M.; Sengupta, P.; Lippincott-Schwartz, J.; Verkhusha, V.V. Photocontrollable fluorescent proteins for superresolution imaging. Annu. Rev. Biophys. 2014, 43, 303–329. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Neil, S.J.D.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 vpu. Nature 2008, 451, 425. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Müller, B.; Hell, S.W.; Kräusslich, H.-G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.; Baumgartel, V.; Ivanchenko, S.; Torrano, A.A.; Brauchle, C.; Muller, B.; Lamb, D.C. Super-resolution imaging of escrt-proteins at HIV-1 assembly sites. PLoS Pathog. 2015, 11, e1004677. [Google Scholar] [CrossRef] [PubMed]
- Van Engelenburg, S.B.; Shtengel, G.; Sengupta, P.; Waki, K.; Jarnik, M.; Ablan, S.D.; Freed, E.O.; Hess, H.F.; Lippincott-Schwartz, J. Distribution of escrt machinery at HIV assembly sites reveals virus scaffolding of escrt subunits. Science 2014, 343, 653. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Heilemann, M. Shedding new light on viruses: Super-resolution microscopy for studying human immunodeficiency virus. Trends Microbiol. 2013, 21, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Sakin, V.; Paci, G.; Lemke Edward, A.; Müller, B. Labeling of virus components for advanced, quantitative imaging analyses. FEBS Lett. 2016, 590, 1896–1914. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.R.; Campbell, R.E.; Gross, L.A.; Martin, B.R.; Walkup, G.K.; Yao, Y.; Llopis, J.; Tsien, R.Y. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: Synthesis and biological applications. J. Am. Chem. Soc. 2002, 124, 6063–6076. [Google Scholar] [CrossRef] [PubMed]
- Nikić, I.; Plass, T.; Schraidt, O.; Szymański, J.; Briggs John, A.G.; Schultz, C.; Lemke Edward, A. Minimal tags for rapid dual-color live-cell labeling and super-resolution microscopy. Angew. Chem. Int. Ed. 2014, 53, 2245–2249. [Google Scholar] [CrossRef] [PubMed]
- Nienhaus, K.; Nienhaus, G.U. Fluorescent proteins for live-cell imaging with super-resolution. Chem. Soc. Rev. 2014, 43, 1088–1106. [Google Scholar] [CrossRef] [PubMed]
- Wiedenmann, J.; Oswald, F.; Nienhaus, G.U. Fluorescent proteins for live cell imaging: Opportunities, limitations, and challenges. IUBMB Life 2009, 61, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Cheong, F.C.; Rémi Dreyfus, B.S.; Amato-Grill, J.; Xiao, K.; Dixon, L.; Grier, D.G. Flow visualization and flow cytometry with holographic video microscopy. Opt. Express 2009, 17, 13071–13079. [Google Scholar] [CrossRef] [PubMed]
- Crocker, J.C.; Grier, D.G. Methods of digital video microscopy for colloidal studies. J. Colloid Interface Sci. 1996, 179, 298–310. [Google Scholar] [CrossRef]
- Henriques, R.; Lelek, M.; Fornasiero, E.F.; Valtorta, F.; Zimmer, C.; Mhlanga, M.M. Quickpalm: 3d real-time photoactivation nanoscopy image processing in imagej. Nat. Methods 2010, 7, 339. [Google Scholar] [CrossRef] [PubMed]
- Holden, S.J.; Uphoff, S.; Kapanidis, A.N. Daostorm: An algorithm for high- density super-resolution microscopy. Nat. Methods 2011, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Schrimpf, W.; Barth, A.; Hendrix, J.; Lamb, D.C. Pam: A framework for integrated analysis of imaging, single-molecule, and ensemble fluorescence data. Biophys. J. 2018, 114, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Shim, S.-H.; He, J.; Zhuang, X. Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods 2011, 8, 499. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Gillette, J.M.; Lippincott-Schwartz, J. Single-particle tracking photoactivated localization microscopy for mapping single-molecule dynamics. Methods Enzymol. 2010, 475, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Modality | Quantities | Range |
|---|---|---|---|
| Fluorescence correlation spectroscopy (FCS) | Confocal microscope, no scanning | Diffusion coefficient | 1–1000 µm2/s |
| Molecular concentration | 1–1000 nM | ||
| Rel. molecular mass (aqueous buffer) | 0.5–1000 kDa | ||
| Stoichiometry | any, if monodisperse | ||
| Dissociation constant | 1–1000 nM | ||
| Raster image correlation spectroscopy (RICS) | CLSM, PIE-CLSM, scanning disk (SD)-CLSM | Diffusion coefficient | 1–1000 µm2/s |
| Molecular concentration | 1–1000 nM | ||
| Rel. molecular mass (aqueous buffer) | 0.5–1000 kDa | ||
| Stoichiometry | any, if monodisperse | ||
| Dissociation constant | 1–1000 nM | ||
| Temporal image correlation spectroscopy (TICS) | CLSM, PIE-CLSM, TIRFM | Diffusion coefficient | 0.001–10 µm2/s |
| Molecular concentration | 1–1000 nM | ||
| Stoichiometry | any, if monodisperse | ||
| Dissociation constant | 1–1000 nM | ||
| Single particle tracking (SPT) | TIRFM, SD-CLSM | Diffusion coefficient | 10−5–10 µm2/s |
| Dual-color SPT | |||
| Cross-correlation (fluorescence cross correlation spectroscopy (FCCS), TICCS, ccRICS) | Confocal, CLSM PIE-Confocal, PIE-CLSM | Stoichiometry | any, if monodisperse |
| Diffusion coefficient | 1–1000 µm2/s (FCCS, ccRICS), 0.001–10 µm2/s (TICCS) | ||
| Binding constant | nM to µM | ||
| Förster resonance energy transfer (FRET) | Wide-field | Molecular distance | 1–10 nm |
| Single-molecule FRET (smFRET) | TIRFM, Confocal, CLSM, PIE-Confocal, PIE-CLSM | Structure | 1 Å precision |
| Photo-activation localization microscopy (PALM)/stochastic optical reconstruction microscopy (STORM) | Wide-field, TIRFM | Structure information | 20–30 nm precision 20–30 nm precision |
| Colocalization | |||
| Number and brightness (N&B) | Confocal, CLSM PIE-Confocal, PIE-CLSM | Molecular concentration | 1–1000 nM |
| Stoichiometry | any, if monodisperse |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parveen, N.; Borrenberghs, D.; Rocha, S.; Hendrix, J. Single Viruses on the Fluorescence Microscope: Imaging Molecular Mobility, Interactions and Structure Sheds New Light on Viral Replication. Viruses 2018, 10, 250. https://doi.org/10.3390/v10050250
Parveen N, Borrenberghs D, Rocha S, Hendrix J. Single Viruses on the Fluorescence Microscope: Imaging Molecular Mobility, Interactions and Structure Sheds New Light on Viral Replication. Viruses. 2018; 10(5):250. https://doi.org/10.3390/v10050250
Chicago/Turabian StyleParveen, Nagma, Doortje Borrenberghs, Susana Rocha, and Jelle Hendrix. 2018. "Single Viruses on the Fluorescence Microscope: Imaging Molecular Mobility, Interactions and Structure Sheds New Light on Viral Replication" Viruses 10, no. 5: 250. https://doi.org/10.3390/v10050250
APA StyleParveen, N., Borrenberghs, D., Rocha, S., & Hendrix, J. (2018). Single Viruses on the Fluorescence Microscope: Imaging Molecular Mobility, Interactions and Structure Sheds New Light on Viral Replication. Viruses, 10(5), 250. https://doi.org/10.3390/v10050250