Kinetic, Thermodynamic, and Structural Analysis of Drug Resistance Mutations in Neuraminidase from the 2009 Pandemic Influenza Virus

 ,
,
Abstract
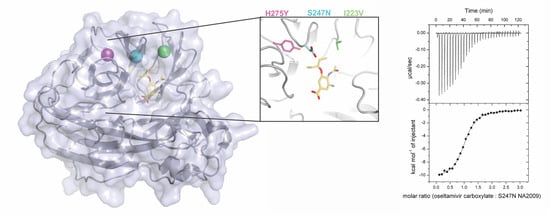
:
1. Introduction
2. Materials and Methods
2.1. Cloning, Expression, and Purification of Recombinant NA2009 Variants
2.2. Enzyme Kinetics of Recombinant NA2009 Variants
2.3. Isothermal Titration Calorimetry
2.4. Protein Crystallography
2.5. Data Collection and Structure Determination
2.6. PDB Accession Codes
3. Results
3.1. The H275Y Mutation Impairs Neuraminidase Activity and Substantially Reduces Susceptibility to Oseltamivir
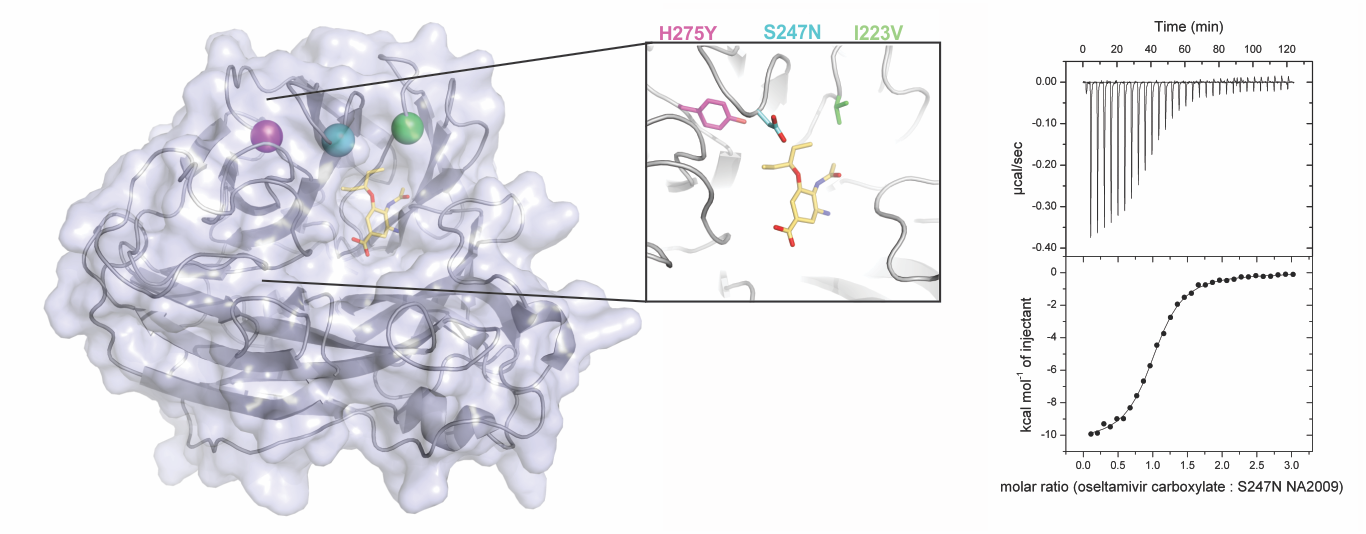
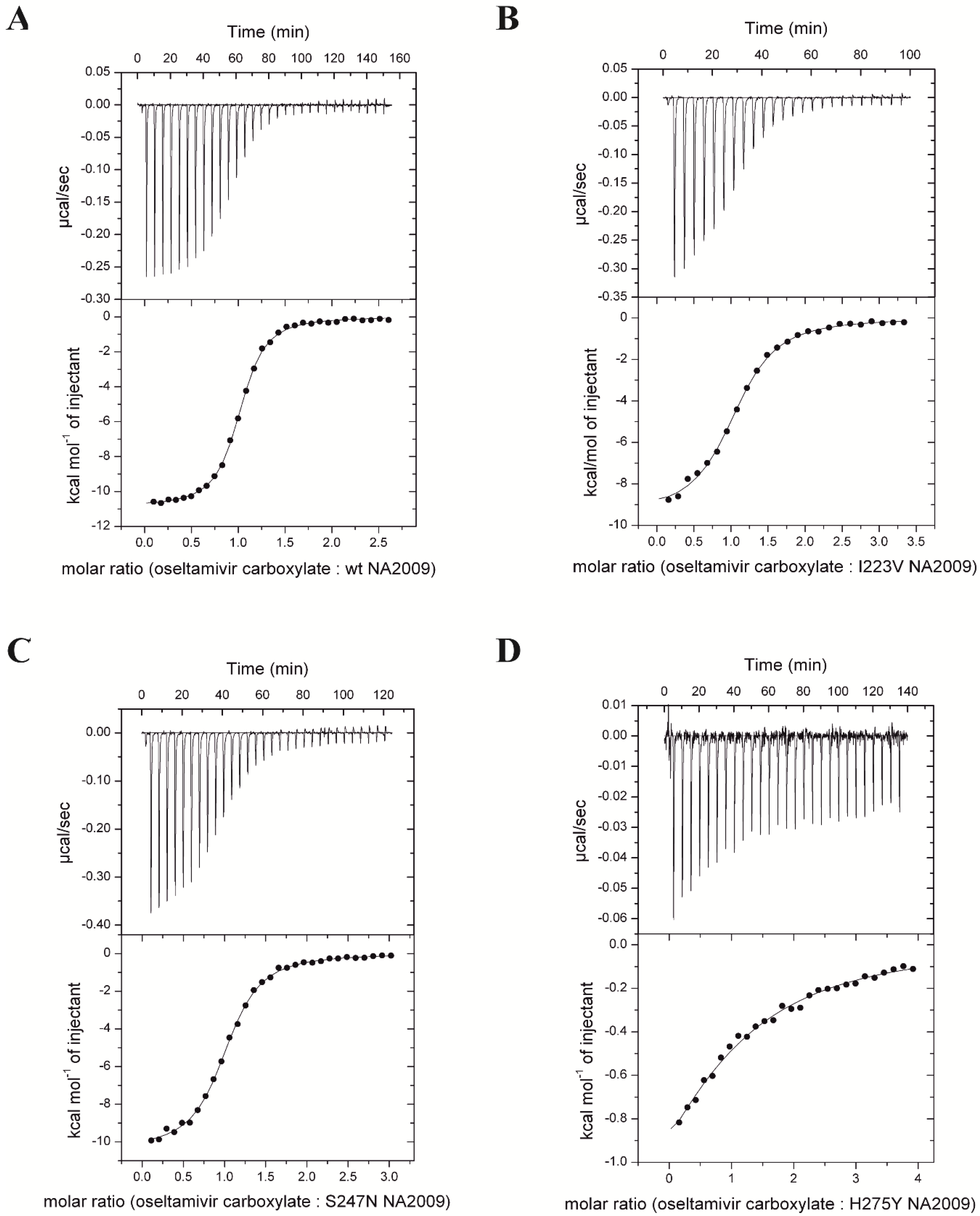
3.2. Neuraminidase Mutations Alter the Thermodynamic Parameters of Oseltamivir Binding
3.3. Crystal Structures Help Elucidate the Mechanism of Oseltamivir Resistance
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klimov, A.I.; Garten, R.; Russell, C.; Barr, I.G.; Besselaar, T.G.; Daniels, R.; Engelhardt, O.G.; Grohmann, G.; Itamura, S.; Kelso, A.; et al. WHO recommendations for the viruses to be used in the 2012 southern hemisphere influenza vaccine: Epidemiology, antigenic and genetic characteristics of influenza A(H1N1)pdm09, A(H3N2) and B influenza viruses collected from February to September 2011. Vaccine 2012, 30, 6461–6471. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: http://www.Who.Int/mediacentre/factsheets/fs211/en/ (accessed on 10 November 2017).
- Murray, C.J.; Lopez, A.D.; Chin, B.; Feehan, D.; Hill, K.H. Estimation of potential global pandemic influenza mortality on the basis of vital registry data from the 1918–1920 pandemic: A quantitative analysis. Lancet 2006, 368, 2211–2218. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of the early isolates of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Pebody, R.; Warburton, F.; Ellis, J.; Andrews, N.; Potts, A.; Cottrell, S.; Johnston, J.; Reynolds, A.; Gunson, R.; Thompson, C.; et al. Effectiveness of seasonal influenza vaccine for adults and children in preventing laboratory-confirmed influenza in primary care in the United Kingdom: 2015/16 end-of-season results. Euro Surveill. 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Osterholm, M.T.; Kelley, N.S.; Sommer, A.; Belongia, E.A. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 36–44. [Google Scholar] [CrossRef]
- Kumar, D.; Michaels, M.G.; Morris, M.I.; Green, M.; Avery, R.K.; Liu, C.; Danziger-Isakov, L.; Stosor, V.; Estabrook, M.; Gantt, S.; et al. Outcomes from pandemic influenza A H1N1 infection in recipients of solid-organ transplants: A multicentre cohort study. Lancet Infect. Dis. 2010, 10, 521–526. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Ruigrok, R.W.; Cusack, S. The 2.2 A resolution crystal structure of influenza B neuraminidase and its complex with sialic acid. EMBO J. 1992, 11, 49–56. [Google Scholar] [PubMed]
- Varghese, J.N.; Mckimmbreschkin, J.L.; Caldwell, J.B.; Kortt, A.A.; Colman, P.M. The structure of the complex between influenza-virus neuraminidase and sialic-acid, the viral receptor. Proteins 1992, 14, 327–332. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. A revision of the system of nomenclature for influenza viruses: A WHO memorandum. Bull. World Health Organ. 1980, 58, 585–591. [Google Scholar]
- Prachanronarong, K.L.; Ozen, A.; Thayer, K.M.; Yilmaz, L.S.; Zeldovich, K.B.; Bolon, D.N.; Kowalik, T.F.; Jensen, J.D.; Finberg, R.W.; Wang, J.P.; et al. Molecular basis for differential patterns of drug resistance in influenza N1 and N2 neuraminidase. J. Chem. Theory Comput. 2016, 12, 6098–6108. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.J.; Haire, L.F.; Stevens, D.J.; Collins, P.J.; Lin, Y.P.; Blackburn, G.M.; Hay, A.J.; Gamblin, S.J.; Skehel, J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Oxford, J.S. Chemoprophylaxis and Virus Infections of the Respiratory Tract; CRC Press: Cleveland, OH, USA, 1977; Volume 1, pp. 140–187. [Google Scholar]
- Colman, P.M. Neuraminidase inhibitors as antivirals. Vaccine 2002, 20, S55–S58. [Google Scholar] [CrossRef]
- Yang, X.Y.; Steukers, L.; Forier, K.; Xiong, R.H.; Braeckmans, K.; Van Reeth, K.; Nauwynck, H. A beneficiary role for neuraminidase in influenza virus penetration through the respiratory mucus. PLoS ONE 2014, 9, e110026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Itzstein, M.; Wu, W.Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.U.; Lew, W.; Williams, M.A.; Liu, H.; Zhang, L.; Swaminathan, S.; Bischofberger, N.; Chen, M.S.; Mendel, D.B.; Tai, C.Y.; et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997, 119, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Babu, Y.S.; Chand, P.; Bantia, S.; Kotian, P.; Dehghani, A.; El-Kattan, Y.; Lin, T.H.; Hutchison, T.L.; Elliott, A.J.; Parker, C.D.; et al. Bcx-1812 (rwj-270201): Discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J. Med. Chem. 2000, 43, 3482–3486. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Chang, S.C.; Kim, M.J.; Chu, D.W.; Ohashi, Y. Long-acting neuraminidase inhibitor laninamivir octanoate versus oseltamivir for treatment of influenza: A double-blind, randomized, noninferiority clinical trial. Clin. Infect. Dis. 2010, 51, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Varghese, J.N.; Laver, W.G.; Colman, P.M. Structure of the influenza-virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature 1983, 303, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Colman, P.M.; Varghese, J.N.; Laver, W.G. Structure of the catalytic and antigenic sites in influenza-virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Varghese, J.N.; Smith, P.W.; Sollis, S.L.; Blick, T.J.; Sahasrabudhe, A.; McKimm-Breschkin, J.L.; Colman, P.M. Drug design against a shifting target: A structural basis for resistance to inhibitors in a variant of influenza virus neuraminidase. Structure 1998, 6, 735–746. [Google Scholar] [CrossRef]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.; Priyamvada, L.; Ende, Z.; Steel, J.; Lowen, A.C. Influenza virus reassortment occurs with high frequency in the absence of segment mismatch. PLoS Pathog. 2013, 9, e1003421. [Google Scholar] [CrossRef] [PubMed]
- Ives, J.A.L.; Carr, J.A.; Mendel, D.B.; Tai, C.Y.; Lambkin, R.; Kelly, L.; Oxford, J.S.; Hayden, F.G.; Roberts, N.A. The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antivir. Res. 2002, 55, 307–317. [Google Scholar] [CrossRef]
- Meijer, A.; Lackenby, A.; Hungnes, O.; Lina, B.; van-der-Werf, S.; Schweiger, B.; Opp, M.; Paget, J.; van-de-Kassteele, J.; Hay, A.; et al. Oseltamivir-resistant influenza virus A (H1N1), Europe, 2007–08 season. Emerg. Infect. Dis. 2009, 15, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Gong, L.I.; Baltimore, D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 2010, 328, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Abed, Y.; Baz, M.; Boivin, G. Impact of neuraminidase mutations conferring influenza resistance to neuraminidase inhibitors in the N1 and N2 genetic backgrounds. Antivir. Ther. 2006, 11, 971–976. [Google Scholar] [PubMed]
- Collins, P.J.; Haire, L.F.; Lin, Y.P.; Liu, J.; Russell, R.J.; Walker, P.A.; Skehel, J.J.; Martin, S.R.; Hay, A.J.; Gamblin, S.J. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature 2008, 453, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Vergara-Jaque, A.; Poblete, H.; Lee, E.H.; Schulten, K.; Gonzalez-Nilo, F.; Chipot, C. Molecular basis of drug resistance in A/H1N1 virus. J. Chem. Inf. Model. 2012, 52, 2650–2656. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.J.; Malaisree, M.; Long, B.; McIntosh-Smith, S.; Mulholland, A.J. Analysis and assay of oseltamivir-resistant mutants of influenza neuraminidase via direct observation of drug unbinding and rebinding in simulation. Biochemistry 2013, 52, 8150–8164. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, A.; Abed, Y.; Bouhy, X.; Beaulieu, E.; Mallett, C.; Russell, R.; Boivin, G. Impact of mutations at residue I223 of the neuraminidase protein on the resistance profile, replication level, and virulence of the 2009 pandemic influenza virus. Antimicrob. Agents Chemother. 2012, 56, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Fry, A.M.; Gubareva, L.V. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir. Ther. 2012, 17, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Hurt, A.C.; Lee, R.T.; Leang, S.K.; Cui, L.; Deng, Y.M.; Phuah, S.P.; Caldwell, N.; Freeman, K.; Komadina, N.; Smith, D.; et al. Increased detection in Australia and Singapore of a novel influenza A(H1N1) 2009 variant with reduced oseltamivir and zanamivir sensitivity due to a S247N neuraminidase mutation. Euro Surveill. 2011, 16, 2–7. [Google Scholar]
- Seibert, C.W.; Rahmat, S.; Krammer, F.; Palese, P.; Bouvier, N.M. Efficient transmission of pandemic H1N1 influenza viruses with high-level oseltamivir resistance. J. Virol. 2012, 86, 5386–5389. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.G.; Pinilla, L.T.; Holder, B.P.; Abed, Y.; Boivin, G.; Beauchemin, C.A.A. Impact of the H275Y and I223V mutations in the neuraminidase of the 2009 pandemic influenza virus in vitro and evaluating experimental reproducibility. PLoS ONE 2015, 10, e0126115. [Google Scholar] [CrossRef] [PubMed]
- McKimm-Breschkin, J.L. Influenza neuraminidase inhibitors: Antiviral action and mechanisms of resistance. Influenza Other Respir. 2013, 7, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, P.; Bank, C.; Renzette, N.; Prachanronarong, K.; Yilmaz, L.S.; Caffrey, D.R.; Zeldovich, K.B.; Schiffer, C.A.; Kowalik, T.F.; et al. A balance between inhibitor binding and substrate processing confers influenza drug resistance. J. Mol. Biol. 2016, 428, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Cao, Y.; Zhou, J.; Qin, K.; Zhu, W.; Zhu, Y.; Yang, L.; Wang, D.; Wei, H.; Shu, Y. A conformational restriction in the influenza a virus neuraminidase binding site by R152 results in a combinational effect of I222T and H274Y on oseltamivir resistance. Antimicrob. Agents Chemother. 2014, 58, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Van der Vries, E.; Veldhuis Kroeze, E.J.; Stittelaar, K.J.; Linster, M.; Van der Linden, A.; Schrauwen, E.J.; Leijten, L.M.; van Amerongen, G.; Schutten, M.; Kuiken, T.; et al. Multidrug resistant 2009 A/H1N1 influenza clinical isolate with a neuraminidase I223R mutation retains its virulence and transmissibility in ferrets. PLoS Pathog. 2011, 7, e1002276. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Pizzorno, A.; Abed, Y.; Boivin, G. Influenza virus resistance to neuraminidase inhibitors. Antivir. Res. 2013, 98, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Itzstein, M. Influenza Virus Sialidase—A Drug Discovery Target; Springer: Basel, Switzerland, 2012. [Google Scholar]
- Potier, M.; Mameli, L.; Belisle, M.; Dallaire, L.; Melancon, S.B. Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-alpha-d-n-acetylneuraminate) substrate. Anal. Biochem. 1979, 94, 287–296. [Google Scholar] [CrossRef]
- Barinka, C.; Rinnova, M.; Sacha, P.; Rojas, C.; Majer, P.; Slusher, B.S.; Konvalinka, J. Substrate specificity, inhibition and enzymological analysis of recombinant human glutamate carboxypeptidase II. J. Neurochem. 2002, 80, 477–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.G.; Skerra, A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007, 2, 1528–1535. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.G.M.; Batz, L.; Bonet, L.; Carl, U.; Holzapfel, G.; Kiem, K.; Matulewicz, K.; Niermeier, D.; Schuchardt, I.; Stanar, K. Development of the Twin-Strep-tag® and its application for purification of recombinant proteins from cell culture supernatants. Protein Expr. Purif. 2013, 92, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Morrison, J.F. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 1979, 63, 437–467. [Google Scholar] [PubMed]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukada, H.; Takahashi, K. Enthalpy and heat capacity changes for the proton dissociation of various buffer components in 0.1 M potassium chloride. Proteins 1998, 33, 159–166. [Google Scholar] [CrossRef]
- Mueller, U.; Darowski, N.; Fuchs, M.R.; Forster, R.; Hellmig, M.; Paithankar, K.S.; Puhringer, S.; Steffien, M.; Zocher, G.; Weiss, M.S. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 2012, 19, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Krug, M.; Weiss, M.S.; Heinemann, U.; Mueller, U. Xdsapp: A graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 2012, 45, 568–572. [Google Scholar] [CrossRef]
- Brunger, A.T. Free R-value—A novel statistical quantity for assessing the accuracy of crystal-structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 1622–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albinana, C.B.; Machara, A.; Rezacova, P.; Pachl, P.; Konvalinka, J.; Kozisek, M. Kinetic, thermodynamic and structural analysis of tamiphosphor binding to neuraminidase of H1N1 (2009) pandemic influenza. Eur. J. Med. Chem. 2016, 121, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Computational Project. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., 3rd; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawthorne, M.F.; Young, D.C.; Wegner, P.A. Carbametallic boron hydride derivatives. I. Apparent analogs of ferrocene and ferricinium ion. J. Am. Chem. Soc. 1965, 87, 1818–1819. [Google Scholar] [CrossRef]
- Betts, M.J.; Sternberg, M.J. An analysis of conformational changes on protein-protein association: Implications for predictive docking. Protein Eng. 1999, 12, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Rezac, J.; Riley, K.E.; Hobza, P. S66: A well-balanced database of benchmark interaction energies relevant to biomolecular structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.J.; Haire, L.F.; Lin, Y.P.; Liu, J.; Russell, R.J.; Walker, P.A.; Martin, S.R.; Daniels, R.S.; Gregory, V.; Skehel, J.J.; et al. Structural basis for oseltamivir resistance of influenza viruses. Vaccine 2009, 27, 6317–6323. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A.J.; Barrett, S.; Peat, T.S.; Newman, J.; Streltsov, V.A.; Waddington, L.; Saito, T.; Tashiro, M.; McKimm-Breschkin, J.L. Structural and functional basis of resistance to neuraminidase inhibitors of influenza B viruses. J. Med. Chem. 2010, 53, 6421–6431. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Qi, J.; Zhang, W.; Vavricka, C.J.; Shi, Y.; Wei, J.; Feng, E.; Shen, J.; Chen, J.; Liu, D.; et al. The 2009 pandemic H1N1 neuraminidase N1 lacks the 150-cavity in its active site. Nat. Struct. Mol. Biol. 2010, 17, 1266–1268. [Google Scholar] [CrossRef] [PubMed]
- Le, L.; Lee, E.H.; Hardy, D.J.; Truong, T.N.; Schulten, K. Molecular dynamics simulations suggest that electrostatic funnel directs binding of tamiflu to influenza N1 neuraminidases. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| NA2009 Variant | H275Y | I223V | S247N | H275Y/I223V | H275Y/S247N |
|---|---|---|---|---|---|
| PDB Code | 5NWE | 5NZ4 | 5NZE | 5NZF | 5NZN |
| Data collection statistics | |||||
| Space group | P21 | C2221 | C2221 | P21 | P21 |
| Cell parameters (Å; °) | a = 85.47 b = 126.60 c = 96.56 α = γ = 90 β = 93.49 | a = 119.46 b = 137.58 c = 118.61 α = β = γ = 90 | a = 118.67 b = 136.59 c = 119.14 α = β = γ = 90 | a = 85.65 b = 127.22 c = 96.79 α = γ = 90 β = 92.93 | a = 85.78 b = 127.54 c = 96.77 α = γ = 90 β = 93.24 |
| Number of molecules in AU | 4 | 2 | 2 | 4 | 4 |
| Wavelength (Å) | 0.918 | 0.918 | 1.54 | 0.918 | 0.918 |
| Resolution (Å) | 48.2–2.00 (2.12–2.00) | 44.92–1.37 (1.41–1.37) | 38.00–1.69 (1.75–1.69) | 45.18–1.75 (1.81–1.75) | 48.31–1.73 (1.79–1.73) |
| Number of unique reflections | 137,306 (21,721) | 198,814 (19,191) | 101,433 (8959) | 207,181 (20,679) | 214,541 (20,953) |
| Redundancy | 3.0 (3.0) | 3.4 (3.4) | 4.8 (3.6) | 3.4 (3.2) | 3.4 (3.2) |
| Completeness (%) | 98.8 (96.9) | 96.6 (93.9) | 93.9 (83.6) | 99.6 (99.8) | 99.4 (97.3) |
| Rmerge a | 13.9 (62.1) | 9.6 (81.5) | 9.3 (68.2) | 11.4 (73.3) | 11.0 (73.1) |
| Average I/σ(I) | 7.6 (2.0) | 10.5 (1.6) | 11.6 (2.0) | 9.0 (1.8) | 9.4 (1.8) |
| Wilson B (Å2) | 27.11 | 11.9 | 16.6 | 16.6 | 15.7 |
| Refinement statistics | |||||
| Resolution range (Å) | 48.19–2.00 (2.05–2.00) | 44.92–1.37 (1.41–1.37) | 38.00–1.69 (1.75–1.69) | 45.18–1.75 (1.81–1.75) | 48.31–1.73 (1.79–1.73) |
| No. of reflections in working set | 135,202 (9455) | 198,810 (19,192) | 101,425 (8959) | 207,153 (20,678) | 214,496 (20,952) |
| No. of reflections in test set | 2100 (147) | 1989 (192) | 2028 (179) | 2176 (217) | 2253 (220) |
| R value (%) b | 23.6 (28.4) | 14.5 (30.7) | 19.1 (25.8) | 22.0 (29.2) | 23.4 (29.6) |
| Rfree value (%) c | 27.7 (34.3) | 17.0 (33.8) | 21.6 (27.0) | 26.3 (31.9) | 26.7 (32.5) |
| RMSD bond length (Å) | 0.014 | 0.017 | 0.019 | 0.017 | 0.019 |
| RMSD angle (°) | 1.60 | 1.77 | 1.79 | 1.77 | 1.86 |
| Number of atoms in AU | 13,704 | 7384 | 7033 | 13,691 | 13,692 |
| Number of protein atoms in AU | 12,008 | 6067 | 6031 | 12,005 | 12,025 |
| Number of water molecules in AU | 1380 | 1084 | 795 | 1337 | 1382 |
| Mean B value all/protein/waters (Å2) | 23.9/22.7/31.0 | 14.3/11.2/28.6 | 17.7/15.8/28.6 | 19.4/18.1/27.4 | 18.2/17.0/26.0 |
| Ramachandran plot statisticsd | |||||
| Residues in favored regions (%) | 95.5 | 96.2 | 96.8 | 96.2 | 96.0 |
| Residues in allowed regions (%) | 4.4 | 3.8 | 3.2 | 3.6 | 4.0 |
| NA Mutation a | Km [mM] | kcat [s−1] | kcat/Km [M−1 s−1] | Ki [nM] | Fold Ki b |
|---|---|---|---|---|---|
| - | 1.1 ± 0.03 | 0.90 ± 0.01 | 820 ± 30 | 24 ± 4 | 1 |
| H275Y | 1.1 ± 0.2 | 0.054 ± 0.004 | 48 ± 11 | 27,000 ± 2000 | 1100 |
| I223V | 1.2 ± 0.2 | 0.60 ± 0.05 | 510 ± 110 | 700 ± 100 | 29 |
| S247N | 1.1 ± 0.1 | 0.17 ± 0.01 | 160 ± 20 | 450 ± 40 | 19 |
| H275Y/I223V | 1.3 ± 0.3 | 0.045 ± 0.005 | 35 ± 10 | 94,000 ± 7000 | 3900 |
| H275Y/S247N | 3.3 ± 0.5 | 0.10 ± 0.01 | 31 ± 6 | 220,000 ± 4000 | 9000 |
| Stoichiometry | ΔG | ΔH | −T·ΔS | Kd | Fold | |
|---|---|---|---|---|---|---|
| NA Mutation a | Inhib./NA Unit | kcal mol−1 | kcal mol−1 | kcal mol−1 | nM | Kdb |
| oseltamivir carboxylate | ||||||
| - | 1.05 ± 0.05 | −9.4 ± 0.1 | −10.8 ± 0.1 | 1.5 ± 0.2 | 140 ± 20 | 1 |
| I223V | 1.06 ± 0.01 | −8.6 ± 0.1 | −9.4 ± 0.1 | 0.9 ± 0.2 | 520 ± 30 | 4 |
| S247N | 1.02 ± 0.01 | −8.7 ± 0.03 | −10.4 ± 0.1 | 1.7 ± 0.1 | 410 ± 20 | 3 |
| H275Y | 0.96 ± 0.03 | −6.7 ± 0.03 | −2.1 ± 0.04 | −4.6 ± 0.1 | 13,200 ± 600 | 95 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pokorná, J.; Pachl, P.; Karlukova, E.; Hejdánek, J.; Řezáčová, P.; Machara, A.; Hudlický, J.; Konvalinka, J.; Kožíšek, M. Kinetic, Thermodynamic, and Structural Analysis of Drug Resistance Mutations in Neuraminidase from the 2009 Pandemic Influenza Virus. Viruses 2018, 10, 339. https://doi.org/10.3390/v10070339
Pokorná J, Pachl P, Karlukova E, Hejdánek J, Řezáčová P, Machara A, Hudlický J, Konvalinka J, Kožíšek M. Kinetic, Thermodynamic, and Structural Analysis of Drug Resistance Mutations in Neuraminidase from the 2009 Pandemic Influenza Virus. Viruses. 2018; 10(7):339. https://doi.org/10.3390/v10070339
Chicago/Turabian StylePokorná, Jana, Petr Pachl, Elena Karlukova, Jakub Hejdánek, Pavlína Řezáčová, Aleš Machara, Jason Hudlický, Jan Konvalinka, and Milan Kožíšek. 2018. "Kinetic, Thermodynamic, and Structural Analysis of Drug Resistance Mutations in Neuraminidase from the 2009 Pandemic Influenza Virus" Viruses 10, no. 7: 339. https://doi.org/10.3390/v10070339