Abstract
Primary varicella-zoster virus (VZV) infection causes varicella (chickenpox) and the establishment of a lifelong latent infection in ganglionic neurons. VZV reactivates in about one-third of infected individuals to cause herpes zoster, often accompanied by neurological complications. The restricted host range of VZV and, until recently, a lack of suitable in vitro models have seriously hampered molecular studies of VZV latency. Nevertheless, recent technological advances facilitated a series of exciting studies that resulted in the discovery of a VZV latency-associated transcript (VLT) and provide novel insights into our understanding of VZV latency and factors that may initiate reactivation. Deducing the function(s) of VLT and the molecular mechanisms involved should now be considered a priority to improve our understanding of factors that govern VZV latency and reactivation. In this review, we summarize the implications of recent discoveries in the VZV latency field from both a virus and host perspective and provide a roadmap for future studies.
1. Introduction
Most adults worldwide are infected with the neurotropic human alphaherpesvirus, varicella-zoster virus (VZV) [1]. VZV is the causative agent of two distinct diseases: a generalized vesicular skin rash referred to as varicella (chickenpox), and a localized dermatomal skin rash referred to as herpes zoster (HZ; shingles) [2]. Although varicella and HZ were known to be related [3], it was not until 1965 that the British general practitioner, Dr Robert E. Hope-Simpson, suggested that “herpes zoster is a spontaneous manifestation of varicella infection”. This observation was based on a careful examination of ~3500 patients, including 192 HZ cases, who visited his practice over a 16-year period, combined with cautious reading of the available anatomical and epidemiological literature regarding HZ. This led to his famous hypothesis that: “Following the primary infection (chickenpox), virus becomes latent in the sensory ganglia, where it can be reactivated from time to time (herpes zoster)” [4]. Eighteen years later, this hypothesis was proven by Dr Donald Gilden’s crucial discovery of VZV DNA in latently infected human ganglia [5]. Although we have learned much about VZV biology in general in previous decades, the mechanisms underlying VZV latency and reactivation have remained enigmatic, at least in part due to VZV being a human tropic pathogen that does not cause disease in experimental animal models. However, recent insights into the VZV latency program and newly developed in vitro models using human embryonic stem cell (hESC)-derived neurons for studying viral latency and reactivation have now set the stage to unravel the molecular mechanisms that regulate VZV latency. In preparing this review, we sought to be inclusive of as many view points as possible but to consider only human-derived cell- or tissue-based systems (animal models utilised for VZV studies are the subject of another review in this issue). At the outset, we therefore limit this review to only include studies of VZV latency in which there is a demonstrable lack of infectious virus production and/or any observable pathology.
2. Molecular Biology of VZV
2.1. Structure and Genomic Organization of VZV
VZV particles are pleomorphic to spherical in shape, ~150–200 nm in diameter and are composed of three proteinous layers: a nucleocapsid containing the viral double stranded DNA (dsDNA) genome, a tegument layer, consisting of numerous proteins of both viral and host origin surrounding the nucleocapsid, and an envelope comprising a host-derived lipid bilayer inserted with viral glycoproteins facing outwards (Figure 1A,B) [6,7,8]. Upon entry of a VZV virion into the host cell, tegument proteins are released into the newly infected cell, altering the host environment, thereby inhibiting antiviral responses and influencing the fate of the virus program, i.e., a lytic or latent infection (reviewed in ref. [9]). The VZV dsDNA genome is about 125 kilo base pairs (kbp) in size and has a G + C content of 46%. It is composed of two unique segments, termed unique long (UL) and unique short (US), that are flanked by inverted terminal repeat (TR) and internal repeat (IR) structures with high G + C contents (68% for the TRL/IRL and 59% for the IRS/TRS) (Figure 1C) [10,11,12,13,14,15,16,17,18,19]. The very short (~88 bp) TRL and IRL sequences flank the UL region, while the long (7319 bp) IRS and TRS sequences flank the US region. The composite structure generally allows for two isomeric configurations [17] that differ only in terms of whether the US region is inverted. While up to 5% of virions are reported to contain structural isoforms with an inverted UL region [20], their relative scarcity is attributed to a unique DNA sequence at the extreme 5′ end of the UL region that is required for viral DNA cleavage during packaging [21] i.e., these virions may not contain functional genomes. Five regions of the genome contain tandem direct reiterations (R1, R2, R3, R4, and R5) of short repeat sequences, one of which (R4) is located in the IRS/TRS. All except R5 are G + C rich, and all are subject to length and structural polymorphisms that vary both within and between strains [22,23,24,25,26]. Three of these reiterative regions (R1, R2, and R3) are located within the coding portion of VZV genes (open reading frame (ORF) 11, ORF14, and ORF20, respectively) and may therefore exert an effect on protein function. Two copies of R4 are present in the IR and TR, neighboring the origin of replication (OriS), while R5 is located between ORF60 and ORF61.
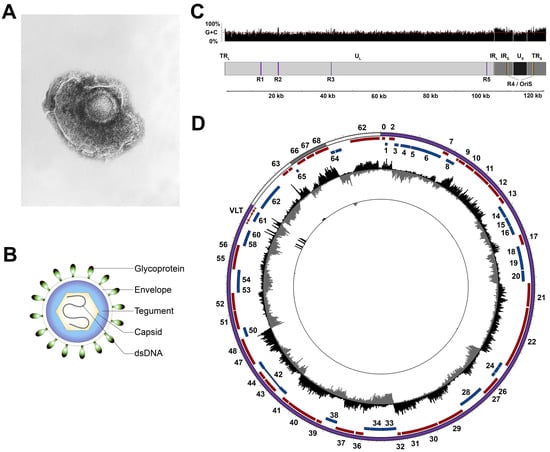
Figure 1.
Structure of varicella-zoster virus (VZV) particles and genome. (A) Electron microscopy image of VZV (obtained from Centers for Disease Control and Prevention (CDC)/Dr Erskine Palmer; B.G. Partin. CDC Public Health Image Library). (B) Schematic representation of the VZV virion. (C) Schematic representation of the VZV genome structure and G + C content. (D) The VZV transcriptome profile during lytic infection of ARPE-19 cells (outer track) and latent infection of human TG (trigeminal ganglia; inner track). Note that the low frequency of VZV-infected neurons in human TG necessitates the use of targeted enrichment of VZV transcripts to detect VZV latency-associated transcript (VLT) expression [27]. Circos plot of the VZV genome (UL, TR/IR, and US are shown as purple, white, and grey bands, respectively; sense and antisense open reading frames (ORFs) are indicated as red and blue blocks, respectively, with ORF numbers indicated where possible). Data represent strand-specific VZV-enriched mRNA-sequencing with peaks facing outwards from the center (black) indicating reads mapping to the sense strand, while peaks facing inward (grey) originate from the antisense strand. The y-axis is scaled to the maximum read depth per library in all cases. dsDNA, double-stranded DNA; UL, unique long; US, unique short; TR, terminal repeat; IR, internal repeat; R, reiterative region; Ori, origin of replication.
2.2. Coding Potential of the VZV Genome
The VZV genome was originally reported to encode 65 unique viral genes, three of which are located in the duplicated IRS/TRS region [19]. Four additional VZV genes have since been identified including ORF0 [28], ORF9A [29], ORF33.5 [30], and the newly discovered VZV latency-associated transcript (VLT) [27] (Figure 1D). An underappreciated feature of VZV is that transcription of several genes, including ORF0, ORF42/45, ORF50, and VLT, require the host-splicing machinery to remove introns from pre-mRNA and have also shown evidence of alternative splicing, resulting in the synthesis of alternative proteins [19,27,31,32,33]. It thus seems likely that the full transcriptional potential of VZV has yet to be revealed, and we predict that the latest technological advances (e.g., full length sequencing of native RNA [34]) will yield further novel discoveries. It is also worth noting that the encoding of additional RNA types, including microRNAs and small non-coding RNAs, is still an area of active study with contrasting results [27,35,36]. How these studies might have an impact on our understanding of viral latency remains to be seen.
2.3. VZV Gene Expression during Productive Infection
Based on limited experimental data [37,38], and mostly by analogy to other alphaherpesviruses, VZV genes have been loosely designated as immediate-early (IE) genes, early (E) genes, and late (L) genes, with each ordered wave of expression thought to be dependent on the protein products of previous classes. Proteins encoded by IE genes act as transcriptional regulators, while those produced by E genes are mainly involved in DNA replication, and L genes encode structural proteins that are crucial for virion formation and egress. VZV encodes at least five transcriptional regulatory proteins specified by four putative IE genes, ORF4, ORF61, ORF62 and ORF63, and one L gene, ORF10. All except the ORF61 protein, IE61, are part of the VZV virion [39,40]. Our understanding of the transcriptional regulation of VZV genes remains incomplete, in part due to the high cell-associated nature of VZV that precludes synchronized infections using cell-free viruses. The dominant transcriptional regulator and possibly only true immediate-early protein encoded by Varicellovirus is homologous to VZV IE62 [39,41,42,43,44,45,46,47,48]. Consistent with this idea, the VZV IE62 major viral transactivator protein can activate all three kinetic classes of VZV genes in the absence of other viral proteins, including all IE genes, ORF4, ORF61, ORF62, and ORF63, while IE4, IE61 and IE63 either do not or minimally stimulate the ORF61 promoter ([49,50,51], reviewed in ref. [52]). Host transcription factors, either by themselves or through interactions with viral transcriptional regulatory proteins, also contribute to viral gene expression (reviewed in ref. [52]). VZV virion proteins delivered into newly infected cells upon entry are not absolutely required to initiate VZV gene expression, as evidenced by the resulting VZV replication upon transfection of cells with viral DNA (reviewed in ref. [53]). Notably, near identical VZV transcriptomes are detected during productive infection of diverse cell types, including neurons [27,54,55,56,57], suggesting a prominent role for either commonly expressed cellular transcription factors or viral proteins in coordinating VZV gene expression.
2.4. Stability of the VZV Genome
Recent advances in the fields of genomics and computational biology have led to an explosion in high-throughput VZV genome sequencing studies [25,58,59,60,61,62,63], with multiple studies focused on exploring the evolution of VZV [60,64,65]. Perhaps the most pertinent observations made by these studies are that (i) the VZV genome is very stable with over 98% sequence conservation between the most distant strain sequences to date, and (ii) the evolutionary history of VZV, like other herpesviruses, is shaped by extensive recombination [60], the latter requiring that two or more different viral genomes occupy the same cell nucleus during viral DNA replication. The live-attenuated nature of the VZV vaccine has enabled comparative analyses of vaccine-induced varicella and HZ isolates, showing that the VZV genome remains highly stable during latency [59,62].
3. Location of Latent VZV
3.1. Sites of VZV Latency
During primary infection, VZV infects and establishes lifelong latency in sensory neurons located in the dorsal root ganglia (DRG) and trigeminal ganglia (TG). While VZV DNA has been detected in various other sensory (geniculate, vestibular, and spiral ganglia) [5,66,67,68] and autonomic ganglia (nodose, enteric, and thoracic sympathetic ganglia) [69,70,71], it remains unclear as to whether the virus can establish latency and reactivate from all of these sites, i.e., infection of these sites may lead to the persistence of only VZV genomes that are not capable of reactivating. The only confirmed sites at which VZV can establish latency and reactivate are the DRG and TG, where the latent viral DNA is maintained as a circular episome in ~2–5% of sensory neurons at a median of 5–7 genome copies per neuron [72,73,74,75]. Additionally, VZV DNA and a restricted number of viral transcripts have been detected in intestinal biopsies of naturally infected and vaccinated individuals [69,76,77,78]. Although in situ detection of VZV DNA or RNA in human enteric neurons is lacking, clinical evidence suggests that VZV reactivation from the enteric nervous system (ENS) could be associated with gastrointestinal dysfunction [79,80,81], possibly resulting from reactivation-induced damage to ENS neurons or virus replication in the gut wall.
3.2. Entry of VZV into the Peripheral Nervous System
Two non-mutually exclusive routes by which VZV may infect sensory ganglion neurons have been proposed (Figure 2A). First, VZV could enter nerve endings innervating the dermis at sites of cutaneous lesions and gain access to ganglia by retrograde axonal transport. This route is supported by the detection of viral antigens in Schwann cells and peripheral nerve axons in the dermis of varicella patients [82], and observations that HZ occurs at the site of varicella vaccine inoculation [83] or the sites most affected by varicella [4]. More recently, VZV infection of axons and retrograde axonal transport to neuronal cell bodies was formally demonstrated in cell culture [84,85], possibly involving fusion between VZV-infected non-neuronal cells and neuronal axons [86]. Notably, nerve endings are located in close proximity to the cutaneous vasculature at the dermal–epidermal junction and in hair follicles [87], supporting a model in which VZV may concurrently infect epidermal or hair follicle keratinocytes and neurons via local cell-to-cell spread. Second, VZV-infected lymphocytes, most likely T-cells, could disseminate the virus to ganglia during varicella-associated viremia. This is supported by detection of VZV DNA in ganglia obtained at autopsy from patients in the prodromal stage of varicella [7]. Moreover, localized injection of live-attenuated VZV vaccine virus results in the establishment of viral latency in bilateral DRGs and enteric ganglia [76]. VZV infects T-cells, prolongs their survival via STAT3 phosphorylation and induction of survivin [88], and modulates their phenotype to induce an activated skin-tropic memory T-cell [89,90,91]. While the prerequisites for T-cell entry into ganglia are unknown, intravenously injected VZV-infected tonsillar T-cells have been shown to transfer the virus to human foetal DRG xenografts implanted under the kidney capsule in severe combined immunodeficiency (SCID) mice [92]. Moreover, T-cells infected with the simian varicella virus (SVV), the closest relative to VZV and natural cause of varicella and HZ in non-human primates, has been detected in the ganglia of non-human primates during primary infection [93]. Thus, the definitive route(s) by which VZV infects ganglionic neurons, with or without cutaneous innervation, needs to be investigated in future studies.
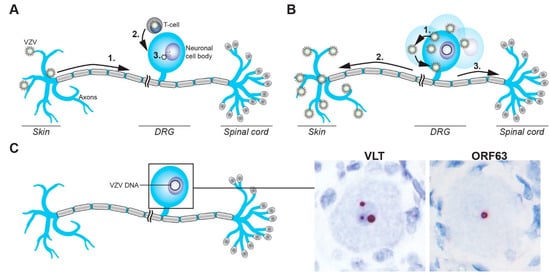
Figure 2.
Schematic representation of the establishment of, and reactivation from, VZV latency in sensory neurons. (A) VZV gains access to sensory ganglion neurons via the infection of nerve endings in skin and retrograde axonal transport to the neuronal cell body (1) or direct infection of cell bodies via VZV-infected T-cells (2), followed by the release of the viral genome into the nucleus (3). (B) VZV reactivation results in virus replication and spread in the cell body (1), followed by transaxonal spread to the skin to cause HZ (2), possibly involving concordant virus spread to the spinal cord (3). (C) VZV latently infected sensory neurons contain viral episomal DNA in their nuclei and express VLT and/or ORF63 RNA, as shown by RNA in situ hybridization on human TG (red signal). Magnification: 400× with 2.5× digital zoom.
4. Transcriptional Repression of Latent VZV Genomes
In the absence of robust animal models, VZV latency studies have been dominated by the use of naturally VZV-infected cadaveric human ganglia, either snap-frozen at autopsy [94] or explanted into short term cultures (reviewed in ref. [95]), although the latter is not reported to result in the production of infectious virions [96]. More recently, the use of in vitro hESC-derived neurons has provided a more accessible approach to latency studies [56,97], principally due to the ability to examine both the establishment of, and reactivation from, latency in sequential fashion over longer periods using mutant viruses and assays that are simply not compatible with wild type, VZV-infected, post-mortem ganglia.
4.1. VZV Transcription in Human Ganglia
The difficulty of obtaining and working with post-mortem human ganglia has resulted in discrepant numbers and identities of viral transcripts, and in some cases, the corresponding proteins have been detected [98,99,100,101,102]. The latter has been confounded by the use of ascites-derived murine and rabbit antibodies that also contain endogenous antibodies that react with human blood type A antigens expressed by sensory neurons [103,104]. Consequently, VZV protein expression in human ganglia appears to be absent or extremely rare and is most likely associated with de-repression of the latent viral genome or in the early stages of reactivation [98,100]. The role of cellular dysregulation following death is undoubtedly a major influence [105], reflected by the post-mortem interval (PMI) which is a major factor in determining the number and identity of viral transcripts present [106]. Most recently, unbiased virus nucleotide-enriched ultra-deep RNA-sequencing confirmed that viral transcription in short-PMI (<9 h post mortem) human ganglia is highly restricted and limited to VLT, frequently accompanied by co-expression of ORF63 RNA, albeit at lower quantities than VLT [27,73,74,75] (Figure 2C). Interestingly, fewer TG neurons express VLT (on average, 0.49%) and ORF63 (0.36%) by in situ hybridization than the reported 2–5% of neurons harboring latent VZV DNA [27], suggesting that not all VZV genomes present in TG neurons are transcriptionally active. Although both VLT and ORF63 RNA retain the potential to be translated during latency, these viral proteins could not be detected in latently infected human ganglia by immunohistochemistry [27]. However, given the very low abundance of VLT and ORF63 RNAs, alternative approaches with higher sensitivity, such as screening for the loading of VLT and ORF63 RNA onto polysomes combined with targeted enrichment for viral RNAs could provide conclusive data. Thus, VZV latency in naturally infected human ganglia is characterized by the exclusive expression of VLT and often, ORF63.
4.2. Comparison of VZV Latency In Vivo and In Vitro
Although naturally VZV-infected, short-PMI, human cadaveric ganglia most closely resemble in vivo VZV latency, ganglia from these individuals are likely to have been latently infected for many decades. In contrast, when using in vitro hESC-derived neuronal models, latency is usually profiled within 14 days. Although transcriptome-wide profiling by RNA-sequencing of in vitro “latent” VZV infections has revealed a marked reduction in viral gene expression, all viral genes are expressed in the absence of detectable levels of viral proteins [56,97]. While limited enrichment for the classical VZV latency-associated ORF63 RNA has been observed, it is not known whether these systems express detectable VLT and if so, whether the latent VLT isoform is transcribed [27,56,97]. This has led to the introduction of new terminology, such as non-productive or quiescent infections, and questions over the relative merits of in vitro models. As a counterpoint, these models have proven to be highly informative of early events during the VZV infection of neurons and the establishment of latency. Moreover, in vitro models remain the only system in which the establishment of latency and subsequent reactivation can be studied, with these studies, as a whole, benefitting increasingly from continual improvements in culture longevity and axonal infection protocols.
4.3. Epigenetic Silencing of the Latent VZV Genome
The molecular mechanisms by which VZV latency is established are largely unknown and much of what is assumed is actually informed by studies of the related herpes simplex virus type 1 (HSV-1). Here, the key concept is that during the traversal of neuronal axons, the genome-containing nucleocapsid is delivered to the nucleus in the absence of functional tegument proteins for IE gene transcription (i.e., VZV ORF10 protein—the homolog of HSV-1 virion protein 16 [VP16]), and this allows loading and maintenance of repressive chromatin upon the viral episome which inhibits viral transcription ([107] and reviewed in ref. [108]). Whether this is a rapid or slow process is not known, although data from VZV in vitro models may provide support for the latter, i.e., viral transcription in newly infected neurons is gradually suppressed over time, eventually leading to the transcriptional profile that we correlate with latency in human ganglia (i.e., the expression of VLT and frequently, ORF63 RNA). Either way, the remarkable switch between lytic and latent transcriptional states is best explained by the assembly of repressive chromatins upon the viral episome. Indeed, studies using late PMI (>9 h post mortem) VZV-latently infected human ganglia have demonstrated euchromatic (H3K9ac) chromatin modifications on promoters of latency-associated ORF63 and ORF62, the latter of which is frequently expressed in late PMI deregulated ganglia [109]. In contrast, promoters of viral genes, ORF14 and ORF36, which are not expressed during latency, nor detected in late-PMI ganglia, have not been associated with H3K9ac [109]. The increase in viral transcription with increasing post-mortem intervals suggests that these studies need to be extended to shorter PMI and genome-wide analyses. Indeed, one might speculate that all but VLT and ORF63 loci would be bound by repressive chromatin, and it will be important to determine whether the VLT locus remains active due to the presence of predicted sequence motifs for flanking chromatin insulators such as CCCTC-binding factor (CTCF), as has been observed for HSV-1 [110].
5. The Varicella Zoster Virus Latency-Associated Transcripts: VLT and ORF63
While all alphaherpesviruses express latency-associated transcripts (LATs), their size, coding potential, functions, and mechanisms of action may vary.
The identification of VLT represents a major advance in studies of VZV latency. VLT is encoded antisense to the ORF61, and it is remarkable to note that all known alphaherpesvirus latency-associated transcripts (LATs) originate from genomic regions encoding infected cell polypeptide 0 (ICP0) homologs (Figure 3). This is indicative of significant evolutionary conservation, although the encoding of LATs and ICP0 homologs antisense to each other makes it harder to assess their individual contributions toward the maintenance of this locus. Notably, VZV appears to be the only alphaherpesvirus that has been analysed in detail to date that consistently expresses an additional latency transcript, ORF63, in human TGs.
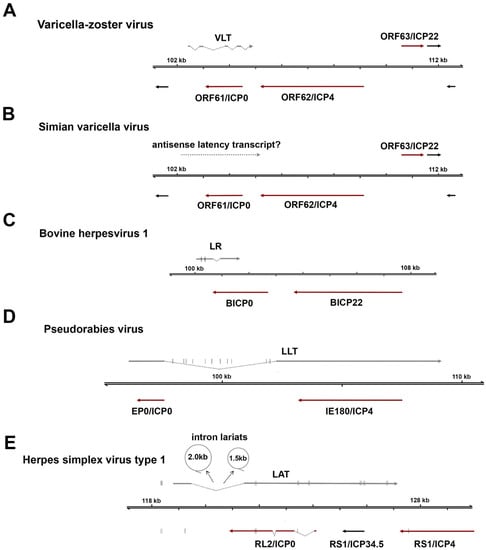
Figure 3.
Comparison of latency-associated transcripts among alphaherpesviruses. All alphaherpesvirus latency-associated transcripts (LATs) are located antisense to the infected cell polypeptide 0 (ICP0) locus, encoding a conserved major immediate-early transactivator (ICP0 or homologs). (A) The VZV latency-associated transcript (VLT) is a 496-nucleotide, multi-exon mRNA that is partially antisense to the ORF61 coding region via exons 3 and 4. (B) Transcripts mapping antisense to simian varicella virus ORF61 are expressed during latency (dotted arrow), but their identity has not yet been defined. (C) Bovine herpesvirus 1 encodes a 2.2 kb latency-related (LR) RNA and encodes two miRNAs in exon 1 (D) The pseudorabies virus large latency transcript (LLT) is the largest characterized alphaherpesvirus latency transcript and encodes eleven distinct miRNAs within the spliced intron. (E) The 8.2 kb herpes simplex virus type 1 (HSV-1) LAT undergoes splicing which yields two highly stable intron lariats, approximately 1.5 and 2.0 kb in size (shown as circles). Note that latency transcripts are shown as grey arrows, immediate early viral transactivators as dark red arrows, and encoded miRNAs as short vertical lines. Scaling across all schematics is equal.
5.1. Comparison of VLT and LATs of Related Alphaherpesviruses
VLT is a polyadenylated RNA comprising at least five distinct exons and is encoded antisense to VZV ORF61, a homolog of the HSV RL2 gene (encoding ICP0). During latency, a single VLT isoform is expressed in the neurons of virtually all analysed VZV-infected human TGs [27], although it is not known whether VLT is expressed in all VZV-infected neurons. VLT appears to be structurally complex compared to the LATs of HSV-1, pseudorabies virus (PRV) and bovine herpesvirus 1 (BHV-1), which are composed of only two exons and a single intron (Figure 3). The possible exception is the closest relative of VZV, the non-human primate SVV, which expresses a transcript mapping to the same region as VLT during latency [111,112], although more detailed studies are needed to determine the exact location and structure of the SVV VLT homolog. Unlike the stable LAT introns of HSV-1 that accumulate to high abundancies in latently infected human TG [27,113], VLT appears to be expressed at relatively few copies per neuron [27]. Furthermore, while the LATs of HSV-1, PRV, and BHV1 encode viral miRNAs [114,115,116], there is no evidence for the expression of VZV miRNAs encoded within VLT—or anywhere else in the VZV genomes—in latently-infected human ganglia [27].
Although VLT is the predominant transcript expressed during latency, its expression is not restricted to the latent phase. During VZV lytic infections of epithelial cell or melanoma cell cultures, multiple alternatively spliced VLT isoforms were identified [27]. Termed VLTly, these lytic isoforms are transcriptionally more complex than the “core” latent VLT isoform detected during latency. Exon skipping, intron retention, and additional upstream exons are all features of VLTly, with most variation apparently occurring between “core” exons 3 and 4. Notably, the core latent VLT isoform has never been observed in lytic infections which may suggest that the core isoform is driven by a cell type (i.e., neuron) specific promoter. During productive infections, VLTly is transcribed with true-late kinetics, is defined as being absolutely dependent on viral replication, and is translated into proteins (pVLT) in cell cultures and zoster skin lesions [27]. Although the expression and function of LATs during lytic alphaherpesvirus infection is incompletely understood, the observed features of VLT are consistent with the general pattern of LATs. During lytic infection, LATs appear to be expressed with late kinetics [27,117]. Alternative LAT isoforms are produced using different transcription start sites or alternative splicing [27,118,119], and some LATs encode proteins [27,120]. Given that hESC neuronal models support both lytic and quiescent VZV infections [56,97], these could be particularly useful for investigating the regulation of lytic and latent isoforms of VLT in the same cell type.
5.2. Function(s) of VLT
The functional characterization of VLT during lytic and latent stages is now a priority. Early investigations using transfection assays showed that VLT selectively represses ORF61 transcription when co-expressed with multiple VZV coding IE genes (ORF61, ORF62 and ORF63) in ARPE-19 epithelial cell cultures, resulting in diminished expression of the ORF61 protein, IE61 [27]. This effect is not pVLT-specific as mutations of the initiating ATG start codon (to ATA) showed a similar effect—suggesting that it is mediated by the RNA itself. While the specific functions and requirements of VLT and VLTly isoforms during lytic and latent infections remain unknown, these data allow us to speculate that VLT may function to maintain latency by repressing the transcription of ORF61, a promiscuous transcriptional regulator, during lytic infections. However, it will be pivotal to confirm these observations in other experimental systems, especially since not all functions attributed to LATs of related herpesviruses in vitro can subsequently be confirmed in vivo [121,122].
5.3. Function(s) of ORF63
VZV expresses two latency-associated transcripts, VLT and ORF63 RNA, the expression of which correlates significantly in human TG, independent of the latent viral DNA load [27], suggesting that expression of both transcripts is linked. It is unclear whether VLT and ORF63 RNA are produced by the same or by distinct populations of neurons, and how this may influence the ability of the virus to reactivate. ORF63 is essential for VZV replication [123] and its encoded protein, IE63, functions not only as a transcriptional regulator activating E genes [124] but also modifies neuronal susceptibility to apoptosis and functions as a immunoevasin that blocks type I interferon (IFN) signaling [125,126,127]. As such, we speculate that ORF63, once translated (IE63), could be crucial for the initiation of reactivation, supported by the correlation between ORF63 RNA abundance and PMI [106]. The observation that IE61 is required for nuclear entry of IE63 in guinea pig enteric neurons, yet to be determined in human neurons [128], suggests that VLT-mediated repression of ORF61 transcription and translation may also function to retain IE63 in the cytoplasm and prevent the transactivation of lytic viral promoters (Figure 4A).
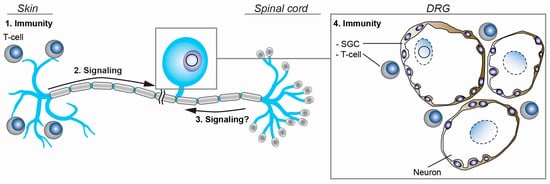
Figure 4.
Schematic representation of the regulation of VZV latency and reactivation. The activation of neuronal signaling pathways in response to stimuli at the periphery (2) and possibly, within the spinal cord (3) may induce VZV reactivation. At the same time, adaptive T-cell-mediated immune responses in skin (1) and ganglia (4), and the local ganglionic innate immunity provided by neuron-interacting satellite glial cells (SGC) are believed to prevent symptomatic VZV reactivation.
6. VZV Reactivation: Restarting Lytic Gene Expression
Symptomatic VZV reactivation, which leads to HZ and/or associated pathologies, typically occurs just once or twice in an infected individual’s lifetime and likely requires two events—stimulus of a latently-infected neuron and the overriding of existing immune protection mechanisms. In contrast, asymptomatic reactivation is thought to occur more frequently [2,129]. Possibly, the phase of intra-ganglionic virus replication that is presumed to occur upon VZV reactivation [130,131] (Figure 2B) before the virus descends down sensory axons to the skin provides the host immune system more time to control most reactivation events before becoming symptomatic. The environmental factors and molecular mechanisms that underpin VZV reactivation in humans remain poorly understood. Clinical observations of HZ after trauma [132] or neurosurgery [133,134] suggest that signalling events from the periphery may induce VZV reactivation in the soma of sensory neurons. Two main signalling pathways have been implicated in VZV reactivation: the phosphatidylinositol-3 kinase (PI3K)-Akt pathway and the mitogen-activated protein kinase (MAPK) pathway. The nerve growth factor (NGF) receptor, TrkA, is expressed by a subpopulation of sensory neurons and signals via the PI3K-Akt and MAPK pathways [135], and depletion of NGF from VZV-latently infected neurons can lead to the reactivation of VZV in some in vitro latency systems [56,97]. The role of NGF signalling in contributing to the maintenance of VZV latency has been confirmed in VZV-latently infected human TG removed within 24 h after death [136]. Intriguingly, the effect of chemical PI3K blockade on VZV reactivation depends on the experimental conditions, as PI3K inhibition did not reactivate VZV in ex vivo cultures of naturally infected human TG neurons [136] and variable effects have been obtained in hESC-derived neurons [56,97]. NGF depletion results in the phosphorylation and activation of the MAPK family member, c-Jun N-terminal kinase (JNK) and is critical for efficient VZV replication in hESC neurons. Selective JNK inhibition limits VZV reactivation in vitro, suggesting JNK signalling plays important role for VZV reactivation in neurons [137].
To reactivate from latency, the latent viral episome needs to be de-repressed, and viral gene expression has to occur as a downstream event of the PI3K-Akt and/or JNK pathways. The mechanisms underlying the reversal of repressive chromatin on the VZV genome and the viral gene expression machinery required for VZV reactivation are poorly understood. Unlike HSV-1, the inhibition of histone deacetylases resulted in failed production of the infectious progeny virus [56], but led to viral DNA replication and expression of late genes in vitro [97]. It may be that additional chromatin modifications, like the methylation of histone H3 lysine 4 (H3K4) or demethylation of H3K9, need to take place before VZV reactivation can occur [138]. Alternatively, JNK signalling may induce a methyl/phospho switch on promoters of VZV lytic genes that facilitates their expression and virus reactivation as observed during HSV-1 latency and reactivation [139]. Clearly, additional environmental factors and neuronal signalling pathways are involved, and the overall outcome of VZV infection will be determined by the combined integration of all these pathways.
7. Intrinsic, Innate, and Adaptive Immunity to VZV Infection in Ganglia
The intrinsic properties of neurons and the innate and adaptive immune responses mounted by ganglion-resident or infiltrating cells that contribute to the control of VZV replication in ganglia are unknown. VZV infection of human foetal DRG xenografts in SCID mice produces transient virus replication and spread, followed by persistently lower levels of viral DNA and limited viral transcription after 4–8 weeks [92], suggesting that adaptive immune responses are not essential for the control of VZV replication in this model system. Intrinsic differences in neuronal subpopulations affect the outcome of VZV infection, as virus replication is blocked in neurons expressing the mechanoreceptive marker RT97, but not in neurons expressing the nociceptive marker peripherin [140]. However, it is not clear which neuron subtype(s) harbor latent VZV in naturally infected humans and whether all neurons are equally capable of supporting VZV reactivation.
7.1. Satellite Glial Cells and Innate Immunity
Although sensory neurons produce little type I IFN α/β, neurons are sensitive to IFN-α/β signalling, as the exposure to IFN-α/β at nerve endings prevents retrograde axonal transport of alphaherpesvirus virions [141]. Notably, various cytokines/chemokines are produced in VZV-infected foetal DRG, including IFN-α, interleukin 1 alpha (IL-1α), IL-6, CXCL10, and transforming growth factor beta [140]. While the cellular source of these cytokines is unknown, satellite glial cells (SGC) are prime candidates. SGC are a specialized ganglionic cell type that shares phenotypic and functional features with professional antigen presenting cells like macrophages and dendritic cells [142,143]. SGC completely enwrap the neuronal cell body and provide physical and nutritional support to the neuron, but also function as tissue-resident innate immune cells that express pattern recognition receptors (e.g., Toll-like receptors), phagocytose, produce inflammatory mediators and potentially regulate local T-cell responses [142,143,144]. Moreover, SGC surrounding HSV-1 infected neurons produce IL-6 and tumour necrosis factor α [145], and we have shown that SGC are activated in response to primary SVV infection in monkeys [146]. However, more detailed studies on the role of SGC in response to ganglionic VZV infected are warranted.
7.2. T-Cell Immunity
VZV reactivation, clinically presenting as HZ, is associated with the waning of systemic VZV-specific T-cell immunity, but not humoral immunity, suggesting a critical role of VZV-specific T-cell memory in preventing virus reactivation [147,148]. However, a large fraction of pathogen-specific memory T-cells are retained in organs, referred to as tissue-resident memory T-cells (TRM), and provide swift protective immunity upon pathogen re-exposure [149,150]. In analogy to HSV-1 mouse models [151,152,153,154] and human TG [155], VZV-specific TRM are expected to home to sites of latency and reactivation in ganglia and skin. Indeed, recent studies showed that VZV-specific TRM cells persist in the healthy skin of latently VZV-infected adults decades after primary infection [156]. Profound T-cell responses detected in ganglia of HZ patients hint at the presence of VZV-specific TRM in ganglia [157,158]. While VZV-specific T-cells in latently infected human TG have not previously been detected, this study analysed only a restricted panel of VZV proteins (IE4, ORF29 protein, IE62, and IE63) [159]. More detailed studies are needed to determine the presence, specificity, and function of VZV-specific TRM in latently VZV-infected human ganglia in regard to their contribution to VZV latency and reactivation control. This will provide more insight into the inverse relationship between VZV-specific T-cell immunity and the risk of HZ development.
8. Future Perspectives
VZV has evolved with the ability to establish lifelong latent infection in neurons, enabling the virus to reactivate later in life and cause recrudescent disease accompanied by VZV spread to naïve individuals. Throughout the decades of latency, the virus must ensure survival of the infected neuron, avoid irreversible silencing of the viral episome, and prevent elimination by host immune responses. Although the mechanisms that coordinate the interplay between the virus and the host remain largely unknown, the many recent exciting advances in the field provide us with the questions to be addressed and tools (ex vivo and in vitro latency models) that can be used to answer them. Thirty-five years on from the discovery of latent VZV residing in sensory ganglia [5], the identification of VLT and the latent VZV transcriptome in naturally-infected, short-PMI TG has provided us with an new perspective on VZV latency [27]. With this advancement, we also propose to enhance the definition of VZV latency as requiring (i) the presence of the viral genome as an episome in the nucleus of host cells without production of infectious progeny, (ii) the capacity of latent VZV to reactivate and produce infectious virus, and (iii) a distinct and restricted pattern of viral gene transcription, i.e., exclusive expression of VLT and/or ORF63 RNA.
It is now critical to determine the roles of (and potential interplay between) VLT and ORF63 RNA in latently infected short-PMI human TG [106] and to analyse their functions in the establishment of quiescence in human pluripotent stem cell (hPSC; hESC/hiPSC)-derived neuronal models [56,97,160,161] or in the reactivation from latency using these same models and/or ex vivo cultures of dissociated naturally VZV-infected human ganglia [136]. Human TG are composed of diverse subtypes of neurons [162], and not all neuronal subpopulations may be equally susceptible to VZV infection [140], as has been shown for HSV-1 [163,164]. Therefore, in situ analyses are required to determine whether VLT and ORF63 RNA are co-expressed by the same or distinct neurons and to define the subtypes of neurons harbouring the latent VZV genome. Recent developments in the generation of recombinant VZV (reviewed in ref. [53]) will facilitate the functional analysis of VLT/pVLT and ORF63 RNA/IE63 using hPSC-derived neuronal systems. Current evidence suggests that both pVLT and IE63 are not expressed, or are expressed below detectable levels, in latently infected ganglia [27,103,104]. While we have identified one possible function of the VLT RNA [27], no functions are currently attributed to ORF63 RNA or pVLT.
Transcriptional regulation of VZV gene expression during lytic and latent infection has been poorly defined. In analogy to HSV-1 [165], the reactivation of latent VZV is presumed to follow a regulated cascade of gene expression, and more detailed analyses of lytic VZV transcriptional regulation are warranted to provide insight into the requirements of VZV reactivation. Recent developments in next-generation sequencing have facilitated the detection of low-abundant VZV genomes in latently infected human TG [58,59] and will open up new avenues to unravel epigenetic modifications of latent VZV genomes using chromatin immunoprecipitation sequencing (ChIP-seq) for histone modifications or CTCF binding. Furthermore, in vitro quiescence models could be used to prospectively map epigenetic chromatin modifications on the VZV genome during quiescent and reactivated stages of infection. Of particular interest is also be the relative expression levels of VLT and ORF63 RNA, and IE63 and IE61 during the initiation of virus reactivation.
The factors triggering and barriers restricting VZV reactivation are poorly understood and are of great interest for future research. The infrequent clinical reactivation of latent VZV suggests that either virus reactivation is rare or that most reactivation events are cleared before HZ can develop. Local immune responses mediated by VZV-specific TRM in skin [156,166] or ganglion, or neuron-interacting SGC [143,144] are likely to be pivotal factors controlling VZV reactivation, e.g., by secreting IFNs to block early reactivation attempts [167]. The recently developed in vitro and ex vivo models to study VZV reactivation [56,97,136,160,161] facilitate systematic analysis of the signalling pathways modulating viral latency, and could be used to investigate the roles of non-neuronal cells and their secreted antiviral factors in this process.
Post-mortem acquired human samples are extremely informative if handled carefully, especially for investigating human-restricted pathogens like VZV, but they are also restrictive in that the resulting datasets only represent a “snapshot” of latency. To overcome this, we hope that technological advances in specific subtypic differentiation methodology for human sensory/sympathetic neurons or organoid culture system from hPSC combined with single cell sequencing methodologies targeting transcriptomes and epigenomes will provide novel ways to examine the dynamics of VZV latency and provide new insight into the molecular mechanisms that control this.
Finally, the discovery of VLT and its potential role in governing VZV latency and/or reactivation also opens up the tantalising possibility of re-engineering current VZV vaccines not to establish latency or reactivate. While a significant amount of work is required to get from here to there, not least determining whether VLT is truly required for latency and/or reactivation, pursuing this long term goal gives new impetus to the VZV research field.
Author Contributions
All authors contributed to the writing, editing and contents of this manuscript.
Funding
This work was supported, in part, by Public Health Services grant AG032958 from the National Institutes of Health (W.J.D.O.) and by the Takeda Science Foundation, Daiichi Sankyo Foundation of Life Science, Japan Society for the Promotion of Science (JSPS KAKENHI JP17K008858) and the Ministry of Education, Culture, Sports, Science and Technology (MEXT KAKENHI JP17H05816, JP16H06429 and JP16K21723) (T.S.).
Acknowledgments
The authors would like to thank Georges Verjans and Angus Wilson for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining Chronic Viral Infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Breuer, J.; Cohen, J.I.; Cohrs, R.J.; Gershon, M.D.; Gilden, D.; Grose, C.; Hambleton, S.; Kennedy, P.G.E.; Oxman, M.N.; et al. Varicella zoster virus infection. Nat. Rev. Dis. Prim. 2015, 1, 15016. [Google Scholar] [CrossRef] [PubMed]
- Weller, T.H.; Witton, H.M.; Bell, E.J. The etiologic agents of varicella and herpes zoster. J. Exp. Med. 1958, 108, 843–868. [Google Scholar] [CrossRef] [PubMed]
- Hope-Simpson, R.E. The Nature of Herpes Zoster: A Long-term Study and a New Hypothesis. Proc. R. Soc. Med. 1965, 58, 9–20. [Google Scholar] [PubMed]
- Gilden, D.H.; Vafai, A.; Shtram, Y.; Becker, Y.; Devlin, M.; Wellish, M. Varicella-zoster virus DNA in human sensory ganglia. Nature 1983, 306, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.A.; Nii, S.; Grose, C. Imaging of the varicella zoster virion in the viral highways: Comparison with herpes simplex viruses 1 and 2, cytomegalovirus, pseudorabies virus, and human herpes viruses 6 and 7. J. Med. Virol. 2003, 70 (Suppl. 1), S103–S110. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Gilden, D. Varicella-Zoster Virus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2015–2057. [Google Scholar]
- Storlie, J.; Maresova, L.; Jackson, W.; Grose, C. Comparative Analyses of the 9 Glycoprotein Genes Found in Wild-Type and Vaccine Strains of Varicella-Zoster Virus. J. Infect. Dis. 2008, 197, S49–S53. [Google Scholar] [CrossRef] [PubMed]
- Penkert, R.R.; Kalejta, R.F. Tegument protein control of latent herpesvirus establishment and animation. Herpesviridae 2011, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, H.; Haines, H.G.; Biswal, N.; Benyesh-Melnick, M. The Characterization of Varicella-zoster Virus DNA. J. Gen. Virol. 1972, 14, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Oakes, J.E.; Iltis, J.P.; Hyman, R.W.; Rapp, F. Analysis by restriction enzyme cleavage of human varicella-zoster virus DNAs. Virology 1977, 82, 353–361. [Google Scholar] [CrossRef]
- Richards, J.C.; Hyman, R.W.; Rapp, F. Analysis of the DNAs from seven varicella-zoster virus isolates. J. Virol. 1979, 32, 812–821. [Google Scholar] [PubMed]
- Zweerink, H.J.; Morton, D.H.; Stanton, L.W.; Neff, B.J. Restriction endonuclease analysis of the DNA from varicella-zoster virus: Stability of the DNA after passage in vitro. J. Gen. Virol. 1981, 55, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Straus, S.E.; Aulakh, H.S.; Ruyechan, W.T.; Hay, J.; Casey, T.A.; Vande Woude, G.F.; Owens, J.; Smith, H.A. Structure of varicella-zoster virus DNA. J. Virol. 1981, 40, 516–525. [Google Scholar] [PubMed]
- Straus, S.E.; Owens, J.; Ruyechan, W.T.; Takiff, H.E.; Casey, T.A.; Vande Woude, G.F.; Hay, J. Molecular cloning and physical mapping of varicella-zoster virus DNA. Proc. Natl. Acad. Sci. USA 1982, 79, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.H.; Dohner, D.E.; Wellinghoff, W.J.; Gelb, L.D. Restriction endonuclease analysis of varicella-zoster vaccine virus and wild-type dnas. J. Med. Virol. 1982, 9, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.M.; Geelen, J.L.; Weststrate, M.W.; Wertheim, P.; van der Noordaa, J. XbaI, PstI, and BglII restriction enzyme maps of the two orientations of the varicella-zoster virus genome. J. Virol. 1981, 39, 390–400. [Google Scholar] [PubMed]
- Dumas, A.M.; Geelen, J.L.M.C.; Maris, W.; van der Noordaa, J. Infectivity and Molecular Weight of Varicella-Zoster Virus DNA. J. Gen. Virol. 1980, 47, 233–235. [Google Scholar] [CrossRef]
- Davison, A.J.; Scott, J.E. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 1986, 67, 1759–1816. [Google Scholar] [CrossRef] [PubMed]
- Kinchington, P.R.; Reinhold, W.C.; Casey, T.A.; Straus, S.E.; Hay, J.; Ruyechan, W.T. Inversion and circularization of the varicella-zoster virus genome. J. Virol. 1985, 56, 194–200. [Google Scholar] [PubMed]
- Kaufer, B.B.; Smejkal, B.; Osterrieder, N. The varicella-zoster virus ORFS/L (ORF0) gene is required for efficient viral replication and contains an element involved in DNA cleavage. J. Virol. 2010, 84, 11661–11669. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Hondo, R.; Taguchi, F.; Yogo, Y. Variation of R1 repeated sequence present in open reading frame 11 of varicella-zoster virus strains. J. Virol. 1988, 62, 1097–1100. [Google Scholar] [PubMed]
- Hondo, R.; Yogo, Y. Strain variation of R5 direct repeats in the right-hand portion of the long unique segment of varicella-zoster virus DNA. J. Virol. 1988, 62, 2916–2921. [Google Scholar] [PubMed]
- Sauerbrei, A.; Zell, R.; Wutzler, P. Analysis of repeat units in the R2 region among different Oka varicella-zoster virus vaccine strains and wild-type strains in Germany. Intervirology 2006, 50, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Gray, E.R.; Kundu, S.; Cooray, S.; Poulsen, A.; Aaby, P.; Breuer, J. Evolution of cocirculating varicella-zoster virus genotypes during a chickenpox outbreak in Guinea-Bissau. J. Virol. 2014, 88, 13936–13946. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Tamura, T.; Miyasaka, K.; Shimizu, A.; Ohashi, N.; Itoh, M. Analysis of numbers of repeated units in R2 region among varicella-zoster virus strains. J. Dermatol. Sci. 2003, 31, 129–133. [Google Scholar] [CrossRef]
- Depledge, D.P.; Ouwendijk, W.J.D.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.M.G.M.; Breuer, J. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef] [PubMed]
- Kemble, G.W.; Annunziato, P.; Lungu, O.; Winter, R.E.; Cha, T.A.; Silverstein, S.J.; Spaete, R.R. Open reading frame S/L of varicella-zoster virus encodes a cytoplasmic protein expressed in infected cells. J. Virol. 2000, 74, 11311–11321. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Williams, M.; Cohen, J.I. Disruption of the varicella-zoster virus dUTPase and the adjacent ORF9A gene results in impaired growth and reduced syncytia formation in vitro. Virology 1997, 234, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Preston, V.G.; Kennard, J.; Rixon, F.J.; Logan, A.J.; Mansfield, R.W.; McDougall, I.M. Efficient herpes simplex virus type 1 (HSV-1) capsid formation directed by the varicella-zoster virus scaffolding protein requires the carboxy-terminal sequences from the HSV-1 homologue. J. Gen. Virol. 1997, 78, 1633–1646. [Google Scholar] [CrossRef] [PubMed]
- Sadaoka, T.; Yanagi, T.; Yamanishi, K.; Mori, Y. Characterization of the varicella-zoster virus ORF50 gene, which encodes glycoprotein M. J. Virol. 2010, 84, 3488–3502. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, T.; Ota, M.; Yamanishi, K.; Mori, Y. Characterization of varicella-zoster virus-encoded ORF0 gene-Comparison of parental and vaccine strains. Virology 2010, 405, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Tyler, S.D.; Carpenter, J.E.; Jackson, W.; Mori, Y.; Arvin, A.M.; Grose, C. The attenuated genotype of varicella-zoster virus includes an ORF0 transitional stop codon mutation. J. Virol. 2012, 86, 10695–10703. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RN A sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Nagel, M.A.; Cohrs, R.J.; Gilden, D.H.; Cullen, B.R. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J. Virol. 2009, 83, 10677–10683. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Golani, L.; Ojha, N.K.; Borodiansky-Shteinberg, T.; Kinchington, P.R.; Goldstein, R.S. Varicella-Zoster Virus Expresses Multiple Small Noncoding RNAs. J. Virol. 2017, 91, e01710-17. [Google Scholar] [CrossRef] [PubMed]
- Reichelt, M.; Brady, J.; Arvin, A.M. The replication cycle of varicella-zoster virus: Analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J. Virol. 2009, 83, 3904–3918. [Google Scholar] [CrossRef] [PubMed]
- Roviš, T.L.; Bailer, S.M.; Pothineni, V.R.; Ouwendijk, W.J.D.; Šimić, H.; Babić, M.; Miklić, K.; Malić, S.; Verweij, M.C.; Baiker, A.; et al. Comprehensive analysis of varicella-zoster virus proteins using a new monoclonal antibody collection. J. Virol. 2013, 87, 6943–6954. [Google Scholar] [CrossRef] [PubMed]
- Kinchington, P.R.; Hougland, J.K.; Arvin, A.M.; Ruyechan, W.T.; Hay, J. The varicella-zoster virus immediate-early protein IE62 is a major component of virus particles. J. Virol. 1992, 66, 359–366. [Google Scholar] [PubMed]
- Kinchington, P.R.; Bookey, D.; Turse, S.E. The transcriptional regulatory proteins encoded by varicella-zoster virus open reading frames (ORFs) 4 and 63, but not ORF 61, are associated with purified virus particles. J. Virol. 1995, 69, 4274–4282. [Google Scholar] [PubMed]
- Perera, L.P.; Mosca, J.D.; Sadeghi-Zadeh, M.; Ruyechan, W.T.; Hay, J. The Varicella-Zoster virus immediate early protein, IE62, can positively regulate its cognate promoter. Virology 1992, 191, 346–354. [Google Scholar] [CrossRef]
- Perera, L.P.; Mosca, J.D.; Ruyechan, W.T.; Hayward, G.S.; Straus, S.E.; Hay, J. A major transactivator of varicella-zoster virus, the immediate-early protein IE62, contains a potent N-terminal activation domain. J. Virol. 1993, 67, 4474–4483. [Google Scholar] [PubMed]
- Tyler, J.T.; Allen, K.E.; Everett, R.D. Mutation of a single lysine residue severely impairs the DNA recognition and regulatory functions of the VZV gene 62 transactivator protein. Nucleic Acids Res. 1994, 22, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Moriuchi, M.; Moriuchi, H.; Straus, S.E.; Cohen, J.I. Varicella-Zoster Virus (VZV) Virion-Associated Transactivator Open Reading Frame 62 Protein Enhances the Infectivity of VZV DNA. Virology 1994. [Google Scholar] [CrossRef] [PubMed]
- Kinchington, P.R.; Turse, S.E. Regulated nuclear localization of the varicella-zoster virus major regulatory protein, IE62. J. Infect. Dis. 1998, 178 (Suppl. 1), S16–S21. [Google Scholar] [CrossRef] [PubMed]
- Ruyechan, W.T.; Peng, H.; Yang, M.; Hay, J. Cellular factors and IE62 activation of VZV promoters. J. Med. Virol. 2003, 70, S90–S94. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Peng, H.; Hay, J.; Ruyechan, W.T. Promoter activation by the varicella-zoster virus major transactivator IE62 and the cellular transcription factor USF. J. Virol. 2006, 80, 7339–7353. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; Che, X.; Sung, P.; Sommer, M.H.; Hay, J.; Arvin, A.M. Mutational analysis of varicella-zoster virus (VZV) immediate early protein (IE62) subdomains and their importance in viral replication. Virology 2016, 492, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sommer, M.; Rajamani, J.; Arvin, A.M. Regulation of the ORF61 Promoter and ORF61 Functions in Varicella-Zoster Virus Replication and Pathogenesis. J. Virol. 2009, 83, 7560–7572. [Google Scholar] [CrossRef] [PubMed]
- Michael, E.J.; Kuck, K.M.; Kinchington, P.R. Anatomy of the varicella-zoster virus open-reading frame 4 promoter. J. Infect. Dis. 1998, 178 (Suppl. 1), S27–S33. [Google Scholar] [CrossRef] [PubMed]
- Kost, R.G.; Kupinsky, H.; Straus, S.E. Varicella-Zoster Virus Gene 63: Transcript Mapping and Regulatory Activity. Virology 1995, 209, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Ruyechan, W.T. Roles of cellular transcription factors in VZV replication. Curr. Top. Microbiol. Immunol. 2010, 342, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. The Varicella-Zoster Virus Genome. Curr. Top. Microbiol. Immunol. 2010, 342, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.L.; Bowlin, J.L.; Cohrs, R.J.; Gilden, D.; Jones, K.L. Comparison of varicella-zoster virus RNA sequences in human neurons and fibroblasts. J. Virol. 2014, 88, 5877–5880. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Waldman Ben-Asher, H.; Kinchington, P.R.; Goldstein, R.S. Cellular Transcriptome Analysis Reveals Differential Expression of Pro- and Antiapoptosis Genes by Varicella-Zoster Virus-Infected Neurons and Fibroblasts. J. Virol. 2014, 88, 7674–7677. [Google Scholar] [CrossRef] [PubMed]
- Sadaoka, T.; Depledge, D.P.; Rajbhandari, L.; Venkatesan, A.; Breuer, J.; Cohen, J.I. In vitro system using human neurons demonstrates that varicella-zoster vaccine virus is impaired for reactivation, but not latency. Proc. Natl. Acad. Sci. USA 2016, 113, E2403–E2412. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Dry, I.R.; Frampton, D.; Singh, M.; Kanda, R.K.; Yee, M.B.; Kellam, P.; Hollinshead, M.; Kinchington, P.R.; O’Toole, E.A.; et al. RNA-seq Analysis of Host and Viral Gene Expression Highlights Interaction between Varicella Zoster Virus and Keratinocyte Differentiation. PLoS Pathog. 2014, 10, e1003896. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Palser, A.L.; Watson, S.J.; Lai, I.Y.C.; Gray, E.R.; Grant, P.; Kanda, R.K.; Leproust, E.; Kellam, P.; Breuer, J. Specific Capture and Whole-Genome Sequencing of Viruses from Clinical Samples. PLoS ONE 2011, 6, e27805. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Kundu, S.; Jensen, N.J.; Gray, E.R.; Jones, M.; Steinberg, S.; Gershon, A.; Kinchington, P.R.; Schmid, D.S.; Balloux, F.; et al. Deep sequencing of viral genomes provides insight into the evolution and pathogenesis of varicella zoster virus and its vaccine in humans. Mol. Biol. Evol. 2014, 31, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Norberg, P.; Depledge, D.P.; Kundu, S.; Atkinson, C.; Brown, J.; Haque, T.; Hussaini, Y.; MacMahon, E.; Molyneaux, P.; Papaevangelou, V.; et al. Recombination of Globally Circulating Varicella Zoster Virus. J. Virol. 2015, 89, 7133–7146. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Brown, J.; Macanovic, J.; Underhill, G.; Breuer, J. Viral Genome Sequencing Proves Nosocomial Transmission of Fatal Varicella. J. Infect. Dis. 2016, 214, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Weinert, L.A.; Depledge, D.P.; Kundu, S.; Gershon, A.A.; Nichols, R.A.; Balloux, F.; Welch, J.J.; Breuer, J. Rates of vaccine evolution show strong effects of latency: Implications for varicella zoster virus epidemiology. Mol. Biol. Evol. 2015, 32, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Taudien, S.; Pfaff, F.; Wutzler, P.; Platzer, M.; Sauerbrei, A. Sequencing of 21 Varicella-Zoster Virus Genomes Reveals Two Novel Genotypes and Evidence of Recombination. J. Virol. 2012, 86, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Tyler, S.D.; Grose, C.; Severini, A.; Gray, M.J.; Upton, C.; Tipples, G.A. A Full-Genome Phylogenetic Analysis of Varicella-Zoster Virus Reveals a Novel Origin of Replication-Based Genotyping Scheme and Evidence of Recombination between Major Circulating Clades. J. Virol. 2006, 80, 9850–9860. [Google Scholar] [CrossRef] [PubMed]
- Tyler, S.D.; Peters, G.A.; Grose, C.; Severini, A.; Gray, M.J.; Upton, C.; Tipples, G.A. Genomic cartography of varicella-zoster virus: A complete genome-based analysis of strain variability with implications for attenuation and phenotypic differences. Virology 2007, 359, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Takasu, T.; Fukuda, S.; Sato-Matsumura, K.C.; Inuyama, Y.; Hondo, R.; Nagashima, K. Detection of varicella-zoster virus DNA in human geniculate ganglia by polymerase chain reaction. J. Infect. Dis. 1992, 166, 1157–1159. [Google Scholar] [CrossRef] [PubMed]
- Gilden, D.H.; Rozenman, Y.; Murray, R.; Devlin, M.; Vafai, A. Detection of varicella-zoster virus nucleic acid in neurons of normal human thoracic ganglia. Ann. Neurol. 1987, 22, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Takasu, T.; Suzuki, S.; Fukuda, S.; Inuyama, Y.; Nagashima, K. Detection of latent varicella-zoster virus infection in human vestibular and spiral ganglia. J. Med. Virol. 1997, 51, 214–216. [Google Scholar] [CrossRef]
- Gershon, A.A.; Chen, J.; Gershon, M.D. A Model of Lytic, Latent, and Reactivating Varicella-Zoster Virus Infections in Isolated Enteric Neurons. J. Infect. Dis. 2008, 197, S61–S65. [Google Scholar] [CrossRef] [PubMed]
- Gilden, D.H.; Gesser, R.; Smith, J.; Wellish, M.; Laguardia, J.J.; Cohrs, R.J.; Mahalingam, R. Presence of VZV and HSV-1 DNA in human nodose and celiac ganglia. Virus Genes 2001, 23, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Nagel, M.A.; Rempel, A.; Huntington, J.; Kim, F.; Choe, A.; Gilden, D. Frequency and abundance of alphaherpesvirus DNA in human thoracic sympathetic ganglia. J. Virol. 2014, 88, 8189–8192. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.; Beer, T.; Cohrs, R.; Gilden, D.H. Configuration of latent varicella-zoster virus DNA. J. Virol. 1995, 69, 8151–8154. [Google Scholar] [PubMed]
- Kennedy, P.G.; Grinfeld, E.; Gow, J.W. Latent varicella-zoster virus is located predominantly in neurons in human trigeminal ganglia. Proc. Natl. Acad. Sci. USA 1998, 95, 4658–4662. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.J.; Cai, G.Y.; Manchak, M.D.; Pizer, L.I. Varicella-zoster virus DNA in cells isolated from human trigeminal ganglia. J. Virol. 2003, 77, 6979–6987. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lau, T.Y.; Morales, M.; Mont, E.K.; Straus, S.E. Laser-Capture Microdissection: Refining Estimates of the Quantity and Distribution of Latent Herpes Simplex Virus 1 and Varicella-Zoster Virus DNA in Human Trigeminal Ganglia at the Single-Cell Level. Cancer Biol. Ther. 2005, 79, 14079–14087. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Chen, J.; Davis, L.; Krinsky, C.; Cowles, R.; Reichard, R.; Gershon, M. Latency of varicella zoster virus in dorsal root, cranial, and enteric Ganglia in vaccinated children. Trans. Am. Clin. Climatol. Assoc. 2012, 123, 17–35. [Google Scholar] [PubMed]
- Chen, J.J.; Gershon, A.A.; Li, Z.; Cowles, R.A.; Gershon, M.D. Varicella zoster virus (VZV) infects and establishes latency in enteric neurons. J. Neurovirol. 2011, 17, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.; van Veen, S.; Mehraban, T.; Mahalingam, R.; Verjans, G. Simian Varicella Virus Infects Enteric Neurons and α4β7 Integrin-Expressing Gut-Tropic T-Cells in Nonhuman Primates. Viruses 2018, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Chen, J.; Gershon, M.D. Use of Saliva to Identify Varicella Zoster Virus Infection of the Gut. Clin. Infect. Dis. 2015, 61, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, M.F.; Salcines-Caviedes, J.R.; Gómez Román, J.; Cano-Hoz, M.; Fernández-Ayala, M.; Casuso-Sáenz, E.; Abascal-Carrera, I.; Campo-Ruiz, A.; Martín, M.C.; Díaz-Pérez, A.; et al. Varicella-zoster virus (VZV) infection as a possible cause of Ogilvie’s syndrome in an immunocompromised host. J. Clin. Microbiol. 2014, 52, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Pui, J.C.; Furth, E.E.; Minda, J.; Montone, K.T. Demonstration of varicella-zoster virus infection in the muscularis propria and myenteric plexi of the colon in an HIV-positive patient with herpes zoster and small bowel pseudo-obstruction (Ogilvie’s syndrome). Am. J. Gastroenterol. 2001, 96, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, P.W.; Lungu, O.; Panagiotidis, C.; Zhang, J.H.; Silvers, D.N.; Gershon, A.A.; Silverstein, S.J. Varicella-zoster virus proteins in skin lesions: Implications for a novel role of ORF29p in chickenpox. J. Virol. 2000, 74, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Hardy, I.; Gershon, A.A.; Steinberg, S.P.; LaRussa, P.; Varicella Vaccine Collaborative Study Group. The Incidence of Zoster after Immunization with Live Attenuated Varicella Vaccine. N. Engl. J. Med. 1991, 325, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Grigoryan, S.; Sloutskin, A.; Yee, M.B.; Zhu, H.; Yang, I.H.; Thakor, N.V.; Sarid, R.; Kinchington, P.R.; Goldstein, R.S. Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: Direct demonstration of axonal infection, transport of VZV, and productive neuronal infection. J. Virol. 2011, 85, 6220–6233. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, S.; Kinchington, P.R.; Yang, I.H.; Selariu, A.; Zhu, H.; Yee, M.; Goldstein, R.S. Retrograde axonal transport of VZV: Kinetic studies in hESC-derived neurons. J. Neurovirol. 2012, 18, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, S.; Yee, M.B.; Glick, Y.; Gerber, D.; Kepten, E.; Garini, Y.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. Direct transfer of viral and cellular proteins from varicella-zoster virus-infected non-neuronal cells to human axons. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.I.; Peltier, A.C.; Li, J. Evaluating dermal myelinated nerve fibers in skin biopsy. Muscle Nerve 2013, 47, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Che, X.; Rajamani, J.; Zerboni, L.; Sung, P.; Ptacek, J.; Arvin, A.M. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.-C.; Padilla, J.A.; Grose, C.; Butcher, E.C.; Arvin, A.M. Tropism of varicella-zoster virus for human tonsillar CD4(+) T lymphocytes that express activation, memory, and skin homing markers. J. Virol. 2002, 76, 11425–11433. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.-C.; Zerboni, L.; Ito, H.; Graham, B.S.; Wallace, M.; Arvin, A.M. Varicella-zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon-alpha. J. Exp. Med. 2004, 200, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Mukherjee, G.; Sen, A.; Bendall, S.C.; Sung, P.; Nolan, G.P.; Arvin, A.M. Single-Cell Mass Cytometry Analysis of Human Tonsil T Cell Remodeling by Varicella Zoster Virus. Cell Rep. 2014, 8, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Ku, C.; Jones, C.D.; Zehnder, J.L.; Arvin, A.M. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6490–6495. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.D.; Mahalingam, R.; de Swart, R.L.; Haagmans, B.L.; van Amerongen, G.; Getu, S.; Gilden, D.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. T-Cell Tropism of Simian Varicella Virus during Primary Infection. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Croen, K.D.; Ostrove, J.M.; Dragovic, L.J.; Straus, S.E. Patterns of gene expression and sites of latency in human nerve ganglia are different for varicella-zoster and herpes simplex viruses. Proc. Natl. Acad. Sci. USA 1988, 85, 9773–9777. [Google Scholar] [CrossRef] [PubMed]
- Steain, M.; Slobedman, B.; Abendroth, A. Experimental models to study varicella-zoster virus infection of neurons. Curr. Top. Microbiol. Immunol. 2010. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Stein, S.; Snyder, M.; Immesoete, P. Attempts to recover varicella virus from ganglia. Ann. Neurol. 1977, 2, 249. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Lebenthal-Loinger, I.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. An In Vitro Model of Latency and Reactivation of Varicella Zoster Virus in Human Stem Cell-Derived Neurons. PLoS Pathog. 2015, 11, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Sobel, R.A.; Ramachandran, V.; Rajamani, J.; Ruyechan, W.; Abendroth, A.; Arvin, A. Expression of varicella-zoster virus immediate-early regulatory protein IE63 in neurons of latently infected human sensory ganglia. J. Virol. 2010, 84, 3421–3430. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, E.; Kennedy, P.G.E. Translation of varicella-zoster virus genes during human ganglionic latency. Virus Genes 2004, 29, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, R.; Wellish, M.; Cohrs, R.; Debrus, S.; Piette, J.; Rentier, B.; Gilden, D.H. Expression of protein encoded by varicella-zoster virus open reading frame 63 in latently infected human ganglionic neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 2122–2124. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.; Grinfeld, E.; Bell, J.E. Varicella-zoster virus gene expression in latently infected and explanted human ganglia. J. Virol. 2000, 74, 11893–11898. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, R.J.; Gilden, D.H.; Kinchington, P.R.; Grinfeld, E.; Kennedy, P.G.E. Varicella-zoster virus gene 66 transcription and translation in latently infected human Ganglia. J. Virol. 2003, 77, 6660–6665. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Sobel, R.A.; Lai, M.; Triglia, R.; Steain, M.; Abendroth, A.; Arvin, A. Apparent expression of varicella-zoster virus proteins in latency resulting from reactivity of murine and rabbit antibodies with human blood group a determinants in sensory neurons. J. Virol. 2012, 86, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.D.; Flowerdew, S.E.; Wick, D.; Horn, A.K.E.; Sinicina, I.; Strupp, M.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M.; Hüfner, K. Immunohistochemical detection of intra-neuronal VZV proteins in snap-frozen human ganglia is confounded by antibodies directed against blood group A1-associated antigens. J. Neurovirol. 2012, 18, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.C.; Mohr, I. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 2012, 20, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.D.; Choe, A.; Nagel, M.A.; Gilden, D.; Osterhaus, A.D.M.E.; Cohrs, R.J.; Verjans, G.M.G.M. Restricted varicella-zoster virus transcription in human trigeminal ganglia obtained soon after death. J. Virol. 2012, 86, 10203–10206. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, W.; Lorentzen, E.U.; Eing, B.R.; Muller, M.; King, N.J.C.; Klupp, B.; Mettenleiter, T.C.; Kuhn, J.E. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog. 2012, 8, e1002679. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, M.P.; Proenca, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef] [PubMed]
- Gary, L.; Gilden, D.H.; Cohrs, R.J. Epigenetic regulation of varicella-zoster virus open reading frames 62 and 63 in latently infected human trigeminal ganglia. J. Virol. 2006, 80, 4921–4926. [Google Scholar] [CrossRef] [PubMed]
- Amelio, A.L.; McAnany, P.K.; Bloom, D.C. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J. Virol. 2006, 80, 2358–2368. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Davis, K.A.; Traina-Dorge, V.; Gray, W.L. Simian varicella virus expresses a latency-associated transcript that is antisense to open reading frame 61 (ICP0) mRNA in neural ganglia of latently infected monkeys. J. Virol. 2007, 81, 8149–8156. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, I.; Barron, A.; Wellish, M.; Engelmann, F.; Legasse, A.; Planer, S.; Gilden, D.; Nikolich-Zugich, J.; Mahalingam, R. Simian varicella virus infection of rhesus macaques recapitulates essential features of varicella zoster virus infection in humans. PLoS Pathog. 2009, 5, e1000657. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.G.; Wagner, E.K.; Devi-Rao, G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 1987, 235, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Jaber, T.; Workman, A.; Jones, C. Small Noncoding RNAs Encoded within the Bovine Herpesvirus 1 Latency-Related Gene Can Reduce Steady-State Levels of Infected Cell Protein 0 (bICP0). J. Virol. 2010, 84, 6297–6307. [Google Scholar] [CrossRef] [PubMed]
- Mahjoub, N.; Dhorne-Pollet, S.; Fuchs, W.; Endale Ahanda, M.-L.; Lange, E.; Klupp, B.; Arya, A.; Loveland, J.E.; Lefevre, F.; Mettenleiter, T.C.; et al. A 2.5-Kilobase Deletion Containing a Cluster of Nine MicroRNAs in the Latency-Associated-Transcript Locus of the Pseudorabies Virus Affects the Host Response of Porcine Trigeminal Ganglia during Established Latency. J. Virol. 2015, 89, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B.; Zhou, G. The 3 facets of regulation of herpes simplex virus gene expression: A. critical inquiry. Virology 2015, 479–480, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Scherba, G. Expression of the pseudorabies virus latency-associated transcript gene during productive infection of cultured cells. J. Virol. 1999, 73, 9781–9788. [Google Scholar] [PubMed]
- Devireddy, L.R.; Jones, C. Alternative splicing of the latency-related transcript of bovine herpesvirus 1 yields RNAs containing unique open reading frames. J. Virol. 1998, 72, 7294–7301. [Google Scholar] [PubMed]
- Sinani, D.; Frizzo da Silva, L.; Jones, C. A Bovine Herpesvirus 1 Protein Expressed in Latently Infected Neurons (ORF2) Promotes Neurite Sprouting in the Presence of Activated Notch1 or Notch3. J. Virol. 2013, 87, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Maillet, S.; Naas, T.; Crepin, S.; Roque-Afonso, A.-M.; Lafay, F.; Efstathiou, S.; Labetoulle, M. Herpes Simplex Virus Type 1 Latently Infected Neurons Differentially Express Latency-Associated and ICP0 Transcripts. J. Virol. 2006, 80, 9310–9321. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Lee, L.Y.; Garber, D.A.; Schaffer, P.A.; Knipe, D.M.; Coen, D.M. Neither LAT nor open reading frame P mutations increase expression of spliced or intron-containing ICP0 transcripts in mouse ganglia latently infected with herpes simplex virus. J. Virol. 2002, 76, 4764–4772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Selariu, A.; Warden, C.; Huang, G.; Huang, Y.; Zaccheus, O.; Cheng, T.; Xia, N.; Zhu, H. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog. 2010, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jackers, P.; Defechereux, P.; Baudoux, L.; Lambert, C.; Massaer, M.; Merville-Louis, M.P.; Rentier, B.; Piette, J. Characterization of regulatory functions of the varicella-zoster virus gene 63-encoded protein. J. Virol. 1992, 66, 3899–3903. [Google Scholar] [PubMed]
- Verweij, M.C.; Wellish, M.; Whitmer, T.; Malouli, D.; Lapel, M.; Jonjić, S.; Haas, J.G.; DeFilippis, V.R.; Mahalingam, R.; Früh, K. Varicella Viruses Inhibit Interferon-Stimulated JAK-STAT Signaling through Multiple Mechanisms. PLoS Pathog. 2015, 11, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Ambagala, A.P.N.; Cohen, J.I. Varicella-Zoster virus IE63, a major viral latency protein, is required to inhibit the alpha interferon-induced antiviral response. J. Virol. 2007, 81, 7844–7851. [Google Scholar] [CrossRef] [PubMed]
- Gerada, C.; Steain, M.; McSharry, B.P.; Slobedman, B.; Abendroth, A. VZV ORF63 protects human neuronal and keratinocyte cell lines from apoptosis and changes its localization upon apoptosis induction. J. Virol. 2018, JVI.00338-18. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.S.; Kyratsous, C.A.; Wan, S.; Silverstein, S. Nuclear import of the varicella-zoster virus latency-associated protein ORF63 in primary neurons requires expression of the lytic protein ORF61 and occurs in a proteasome-dependent manner. J. Virol. 2008, 82, 8673–8686. [Google Scholar] [CrossRef] [PubMed]
- van Velzen, M.; Ouwendijk, W.J.D.; Selke, S.; Pas, S.D.; van Loenen, F.B.; Osterhaus, A.D.M.E.; Wald, A.; Verjans, G.M.G.M. Longitudinal study on oral shedding of herpes simplex virus 1 and varicella-zoster virus in individuals infected with HIV. J. Med. Virol. 2013, 85, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.M.; Tomlinson, A.H. Herpes zoster. Demonstration of virus in trigeminal nerve and ganglion by immunofluorescence and electron microscopy. J. Neurol. Sci. 1972, 15, 35–48. [Google Scholar] [CrossRef]
- Nagashima, K.; Nakazawa, M.; Endo, H. Pathology of the human spinal ganglia in varicella-zoster virus infection. Acta Neuropathol. 1975, 33, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Joesoef, R.M.; Bialek, S.; Wang, C.; Harpaz, R. Association of physical trauma with risk of herpes zoster among medicare beneficiaries in the United States. J. Infect. Dis. 2013, 207, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, E.K.; Brown, C.; Stambough, J.L. Herpes zoster-varicella complicating anterior thoracic surgery: 2 Case reports. J. Spinal Disord. Tech. 2006, 19, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Simms, H.N.; Dunn, L.T. Herpes zoster of the trigeminal nerve following microvascular decompression. Br. J. Neurosurg. 2006, 20, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, J.D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF signaling in sensory neurons: Evidence that early endosomes carry NGF retrograde signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Badani, H.; Baird, N.L.; White, T.M.; Sanford, B.; Gilden, D. Induction of varicella zoster virus DNA replication in dissociated human trigeminal ganglia. J. Neurovirol. 2017, 23, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Kurapati, S.; Sadaoka, T.; Rajbhandari, L.; Shukla, P.; Ali, M.A.; Kim, Y.J.; Lee, G.; Cohen, J.I.; Venkatesan, A. Role of the JNK pathway in varicella-zoster virus lytic infection and reactivation. J. Virol. 2017, 91, e00640-17. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 2009, 15, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Arvin, A. Neuronal Subtype and Satellite Cell Tropism Are Determinants of Varicella-Zoster Virus Virulence in Human Dorsal Root Ganglia Xenografts In Vivo. PLoS Pathog. 2015, 11, e1004989. [Google Scholar] [CrossRef] [PubMed]
- Enquist, L.W.; Leib, D.A. Intrinsic and Innate Defenses of Neurons: Détente with the Herpesviruses. J. Virol. 2017, 91, JVI.01200-16. [Google Scholar] [CrossRef] [PubMed]
- Hanani, M. Satellite glial cells in sensory ganglia: From form to function. Brain Res. Rev. 2005, 48, 457–476. [Google Scholar] [CrossRef] [PubMed]
- van Velzen, M.; Laman, J.D.; Kleinjan, A.; Poot, A.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. Neuron-interacting satellite glial cells in human trigeminal ganglia have an APC phenotype. J. Immunol. 2009, 183, 2456–2461. [Google Scholar] [CrossRef] [PubMed]
- Mitterreiter, J.G.; Ouwendijk, W.J.D.; van Velzen, M.; van Nierop, G.P.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. Satellite glial cells in human trigeminal ganglia have a broad expression of functional Toll-like receptors. Eur. J. Immunol. 2017, 47, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Mori, I.; Goshima, F.; Koshizuka, T.; Imai, Y.; Kohsaka, S.; Koide, N.; Sugiyama, T.; Yoshida, T.; Yokochi, T.; Kimura, Y.; et al. Iba1-expressing microglia respond to herpes simplex virus infection in the mouse trigeminal ganglion. Mol. Brain Res. 2003, 120, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.D.; Getu, S.; Mahalingam, R.; Gilden, D.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. Characterization of the immune response in ganglia after primary simian varicella virus infection. J. Neurovirol. 2016, 22, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.E. Selective decline in cellular immune response to varicella-zoster in the elderly. Neurology 1980, 30, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.J.; Smith, J.G.; Kaufhold, R.M.; Barber, D.; Hayward, A.R.; Chan, C.Y.; Chan, I.S.F.; Li, D.J.J.; Wang, W.; Keller, P.M.; et al. Decline in varicella-zoster virus (VZV)-specific cell-mediated immunity with increasing age and boosting with a high-dose VZV vaccine. J. Infect. Dis. 2003, 188, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; van Gisbergen, K.P.J.M.; Hombrink, P.; van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Palendira, U.; Tscharke, D.C.; Bedoui, S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol. Rev. 2018, 283, 54–76. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Stock, A.T.; Ma, J.Z.; Jones, C.M.; Kent, S.J.; Mueller, S.N.; Heath, W.R.; Carbone, F.R.; Gebhardt, T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl. Acad. Sci. USA 2012, 109, 7037–7042. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, J.E.; Khanna, K.M.; Yee, M.B.; Baty, C.J.; Kinchington, P.R.; Hendricks, R.L. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 2008, 322, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.M.; Bonneau, R.H.; Kinchington, P.R.; Hendricks, R.L. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 2003, 18, 593–603. [Google Scholar] [CrossRef]
- van Velzen, M.; Jing, L.; Osterhaus, A.D.M.E.; Sette, A.; Koelle, D.M.; Verjans, G.M.G.M. Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently infected human trigeminal ganglia. PLoS Pathog. 2013, 9, e1003547. [Google Scholar] [CrossRef] [PubMed]
- Vukmanovic-Stejic, M.; Sandhu, D.; Seidel, J.A.; Patel, N.; Sobande, T.O.; Agius, E.; Jackson, S.E.; Fuentes-Duculan, J.; Suarez-Farinas, M.; Mabbott, N.A.; et al. The Characterization of Varicella Zoster Virus Specific T Cells In Skin and Blood during Ageing Europe PMC Funders Group. J. Investig. Dermatol. 2015, 135, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Steain, M.; Sutherland, J.P.; Rodriguez, M.; Cunningham, A.L.; Slobedman, B.; Abendroth, A. Analysis of T cell responses during active varicella-zoster virus reactivation in human ganglia. J. Virol. 2014, 88, 2704–2716. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, K.; Steain, M.; Cunningham, A.L.; Rodriguez, M.; Blumbergs, P.; Slobedman, B.; Abendroth, A. Characterization of the host immune response in human Ganglia after herpes zoster. J. Virol. 2010, 84, 8861–8870. [Google Scholar] [CrossRef] [PubMed]
- Verjans, G.M.G.M.; Hintzen, R.Q.; van Dun, J.M.; Poot, A.; Milikan, J.C.; Laman, J.D.; Langerak, A.W.; Kinchington, P.R.; Osterhaus, A.D.M.E. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc. Natl. Acad. Sci. USA 2007, 104, 3496–3501. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, T.J.; McCarthy, M.; Cohrs, R.J.; Kaufer, B.B. 3D tissue-like assemblies: A novel approach to investigate virus-cell interactions. Methods 2015, 90, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Sadaoka, T.; Schwartz, C.L.; Rajbhandari, L.; Venkatesan, A.; Cohen, J.I. Human Embryonic Stem Cell-Derived Neurons Are Highly Permissive for Varicella-Zoster Virus Lytic Infection. J. Virol. 2018, 92, JVI-01108. [Google Scholar] [CrossRef] [PubMed]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggström, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Flowerdew, S.E.; Wick, D.; Himmelein, S.; Horn, A.K.E.; Sinicina, I.; Strupp, M.; Brandt, T.; Theil, D.; Hüfner, K. Characterization of Neuronal Populations in the Human Trigeminal Ganglion and Their Association with Latent Herpes Simplex Virus-1 Infection. PLoS ONE 2013, 8, e83603. [Google Scholar] [CrossRef] [PubMed]
- Bertke, A.S.; Patel, A.; Imai, Y.; Apakupakul, K.; Margolis, T.P.; Krause, P.R. Latency-Associated Transcript (LAT) Exon 1 Controls Herpes Simplex Virus Species-Specific Phenotypes: Reactivation in the Guinea Pig Genital Model and Neuron Subtype-Specific Latent Expression of LAT. J. Virol. 2009, 83, 10007–10015. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Mandarino, A.; Chao, M.V.; Mohr, I.; Wilson, A.C. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 2012, 8, e1002540. [Google Scholar] [CrossRef] [PubMed]
- Vukmanovic-Stejic, M.; Sandhu, D.; Sobande, T.O.; Agius, E.; Lacy, K.E.; Riddell, N.; Montez, S.; Dintwe, O.B.; Scriba, T.J.; Breuer, J.; et al. Varicella Zoster Specific CD4 + Foxp3 + T Cells Accumulate after Cutaneous Antigen Challenge in Humans. J. Immunol. 2013, 1, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Linderman, J.A.; Kobayashi, M.; Rayannavar, V.; Fak, J.J.; Darnell, R.B.; Chao, M.V.; Wilson, A.C.; Mohr, I. Immune Escape via a Transient Gene Expression Program Enables Productive Replication of a Latent Pathogen. Cell Rep. 2017, 18, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).