A Reverse Genetics System for Zika Virus Based on a Simple Molecular Cloning Strategy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Antibodies
2.2. Source of Virus Sequences
2.3. In Silico Prediction of CEPs and Sequence Modifications
2.4. Generation of synZIKV Constructs
2.5. In Vitro Transcription and RNA Transfection
2.6. Virus Stocks and Passaging
2.7. Virus Titration by Plaque Assay
2.8. RLuc Assays
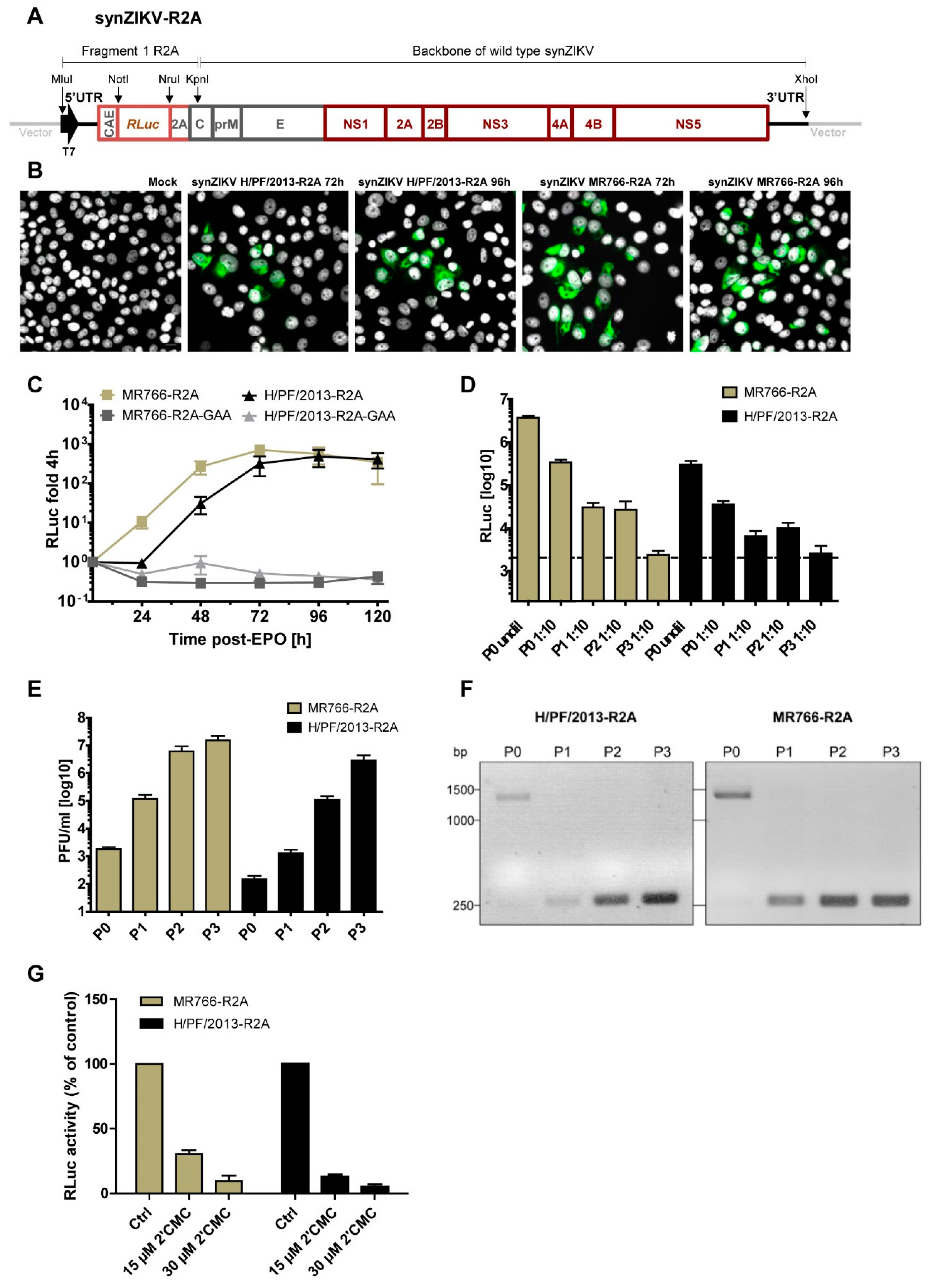
2.9. Antiviral Assays and Stability of synZIKV-R2A Viruses
2.10. Immunofluorescence Microscopy and Western Blotting
3. Results
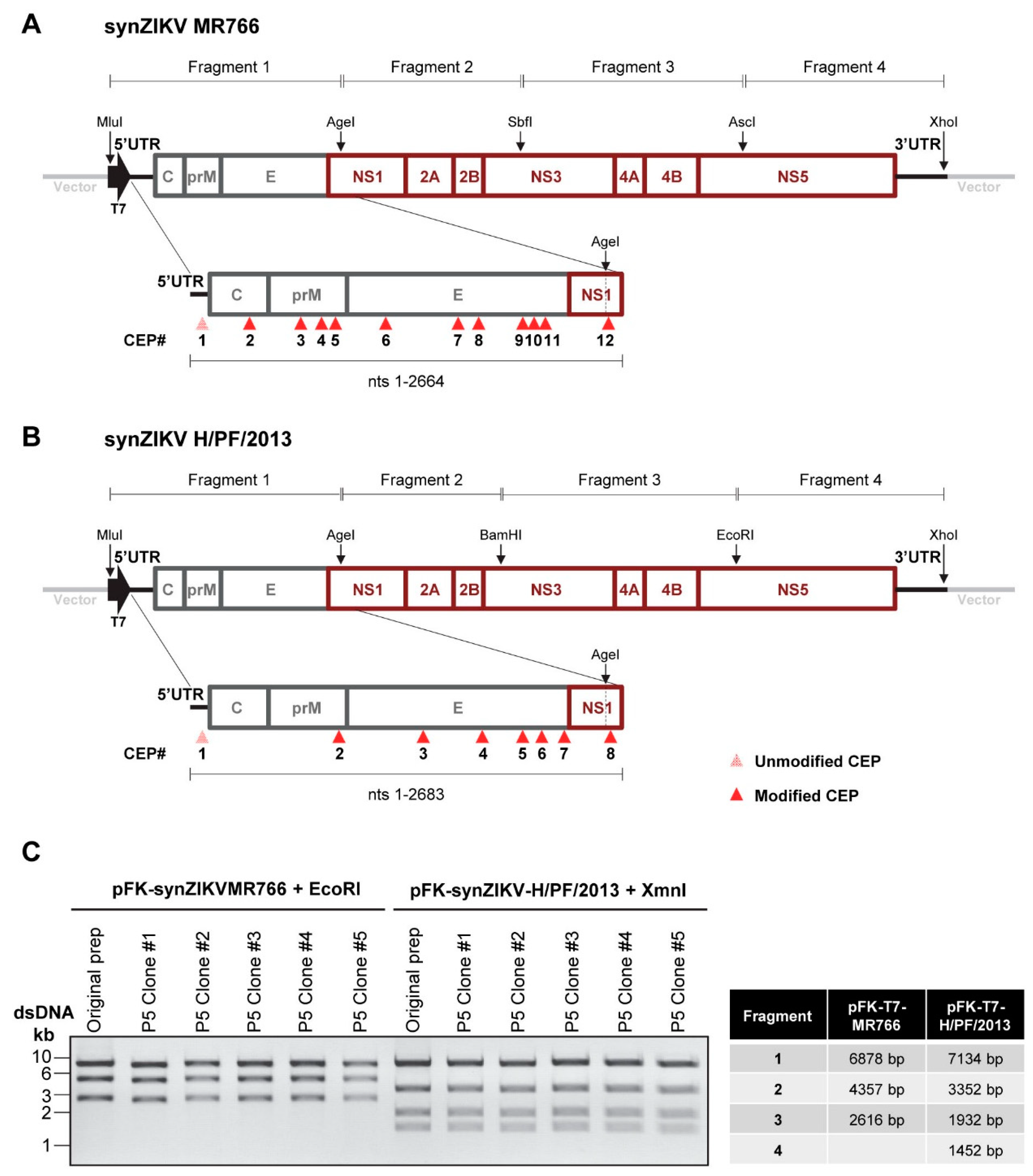
3.1. In Silico Prediction of CEPs and Assembly of Synthetic Full Length ZIKV cDNAs
3.2. Functionality of Full-Length synZIKV Wild-Type Genomes
3.3. Replication and Stability of synZIKV Luciferase Reporter Virus Genomes
3.4. SynZIKV Reporter Viruses Suitable for Live Cell Imaging
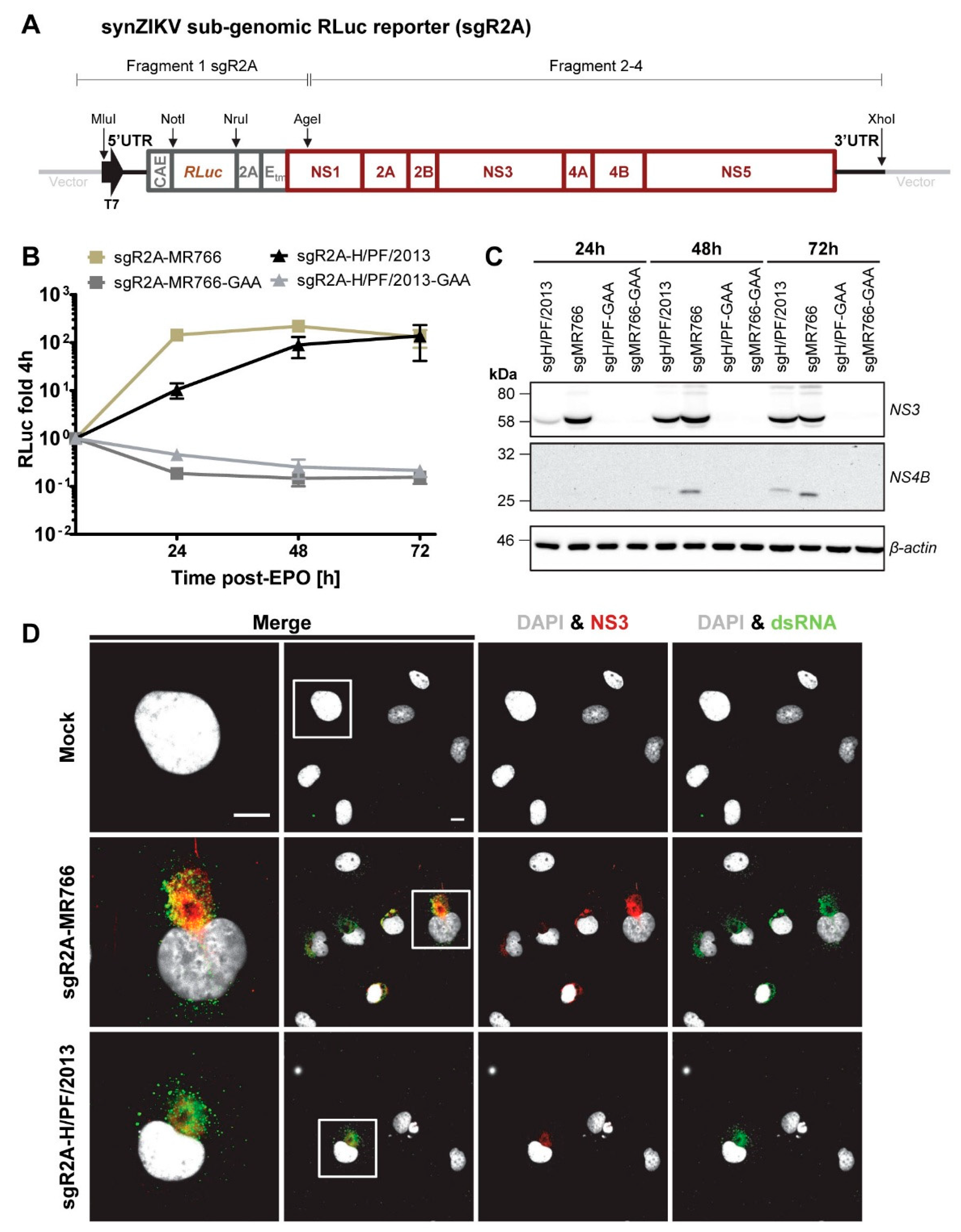
3.5. Sub-Genomic synZIKV Replicons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus (I). Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.H.; Hancock, W.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.M.; Roche, C.; Teissier, A.; Robin, E.; Berry, A.L.; Mallet, H.P.; Sall, A.L.; Musso, D. Zika virus, French Polynesia, South Pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Brasil, P.; Calvet, G.A.; Siqueira, A.M.; Wakimoto, M.; de Sequeira, P.C.; Nobre, A.; de Mendonça, M.C.L.; Lupi, O.; de Souza, R.V.; Romero, C.; et al. Zika Virus Outbreak in Rio de Janeiro, Brazil: Clinical Characterization, Epidemiological and Virological Aspects. PLoS Negl. Trop. Dis. 2016, 10, e0004636. [Google Scholar] [CrossRef] [PubMed]
- Do Rosario, M.S.; de Jesus, P.A.; Vasilakis, N.; Farias, D.S.; Novaes, M.A.; Rodrigues, S.G.; Martins, L.C.; da Costa Vasconcelos, P.F.; Ko, A.I.; Alcântara, L.C., Jr.; et al. Guillain-Barre Syndrome After Zika Virus Infection in Brazil. Am. J. Trop. Med. Hyg. 2016, 95, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Rus, K.R.; Vipotnik, T.V.; Vodušek, V.F.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Haddow, A.D.; Schuh, A.J.; Yasuda, C.Y.; Kasper, M.R.; Heang, V.; Huy, R.; Guzman, H.; Tesh, R.B.; Weaver, S.C. Genetic characterization of Zika virus strains: Geographic expansion of the Asian lineage. PLoS Negl. Trop. Dis. 2012, 6, e1477. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Coyne, C.B. Vaccines in 2017: Closing in on a Zika virus vaccine. Nat. Rev. Immunol. 2018, 18, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Aubry, F.; Nougairede, A.; Gould, E.A.; de Lamballerie, X. Flavivirus reverse genetic systems, construction techniques and applications: A historical perspective. Antivir. Res. 2015, 114, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Fischl, W.; Bartenschlager, R. High-Throughput Screening Using Dengue Virus Reporter Genomes. In Antiviral Methods and Protocols; Humana Press: Totowa, NJ, USA, 2013; Volume 1030, pp. 205–219. [Google Scholar]
- Pu, S.Y.; Wu, R.H.; Yang, C.C.; Jao, T.M.; Tsai, M.H.; Wang, J.C.; Lin, H.M.; Chao, Y.S. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. J. Virol. 2011, 85, 2927–2941. [Google Scholar] [CrossRef] [PubMed]
- Ruggli, N.; Rice, C.M. Functional cDNA Clones of the Flaviviridae: Strategies and Applications. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 1999; Volume 53, pp. 183–207. [Google Scholar]
- Schwarz, M.C.; Sourisseau, M.; Espino, M.M.; Gray, E.S.; Chambers, M.T.; Tortorella, D.; Evans, M.J. Rescue of the 1947 Zika Virus Prototype Strain with a Cytomegalovirus Promoter-Driven cDNA Clone. mSphere 2016, 1, e00246-16. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Kenney, H.; Chen, R.; Liu, G.; Manukyan, H.; Whitehead, S.S.; Laassric, M.; Chumakovc, K.; Pletneva, A.G. A Full-Length Infectious cDNA Clone of Zika Virus from the 2015 Epidemic in Brazil as a Genetic Platform for Studies of Virus-Host Interactions and Vaccine Development. mBio 2016, 7, e01114-16. [Google Scholar] [CrossRef] [PubMed]
- Widman, D.G.; Young, E.; Yount, B.L.; Plante, K.S.; Gallichotte, E.N.; Carbaugh, D.L.; Peck, K.M.; Plante, J.; Swanstrom, J.; Heise, M.T.; et al. A Reverse Genetics Platform That Spans the Zika Virus Family Tree. MBio 2017, 8, e02014-16. [Google Scholar] [CrossRef] [PubMed]
- Weger-Lucarelli, J.; Duggal, N.K.; Bullard-Feibelman, K.; Veselinovic, M.; Romo, H.; Nguyen, C.; Rückert, C.; Brault, A.C.; Bowen, R.A.; Stenglein, M.; et al. Development and Characterization of Recombinant Virus Generated from a New World Zika Virus Infectious Clone. J. Virol. 2017, 91, JVI-01765. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Bos, S.; Krejbich-Trotot, P.; Clain, E.; Viranaicken, W.; El-Kalamouni, C.; Mavingui, P.; Desprès, P. A robust method for the rapid generation of recombinant Zika virus expressing the GFP reporter gene. Virology 2016, 497, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Atieh, T.; Baronti, C.; de Lamballerie, X.; Nougairede, A. Simple reverse genetics systems for Asian and African Zika viruses. Sci. Rep. 2016, 6, 39384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setoh, Y.X.; Prow, N.A.; Peng, N.; Hugo, L.E.; Devine, G.; Hazlewood, J.E.; Suhrbier, A.; Khromykh, A.A. De Novo Generation and Characterization of New Zika Virus Isolate Using Sequence Data from a Microcephaly Case. mSphere 2017, 2, e00190-17. [Google Scholar] [CrossRef] [PubMed]
- Berkeley Drosophila Genome Project, Neural Network Promoter Prediction. Available online: http://www.fruitfly.org/seq_tools/promoter.html (accessed on 15 May 2017).
- Reese, M.G. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 2001, 26, 51–56. [Google Scholar] [CrossRef]
- Acosta, E.G.; Kumar, A.; Bartenschlager, R. Revisiting Dengue Virus-Host Cell Interaction: New Insights into Molecular and Cellular Virology. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2014; Volume 88, pp. 1–109. [Google Scholar]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Schmid, B.; Rinas, M.; Ruggieri, A.; Acosta, E.G.; Bartenschlager, M.; Reuter, A.; Fischl, W.; Harder, N.; Bergeest, J.-P.; Flossdorf, M.; et al. Live Cell Analysis and Mathematical Modeling Identify Determinants of Attenuation of Dengue Virus 2’-O-Methylation Mutant. PLoS Pathog. 2015, 11, e1005345. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell. Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Buhler, S.; Selisko, B.; Davidson, A.; Mulder, K.; Canard, B.; Miller, S.; Bartenschlager, R. Nuclear localization of dengue virus nonstructural protein 5 does not strictly correlate with efficient viral RNA replication and inhibition of type I interferon signaling. J. Virol. 2013, 87, 4545–4557. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Fischl, W.; Scaturro, P.; Cortese, M.; Kallis, S.; Bartenschlager, M.; Fischer, B.; Bartenschlager, R. A Combined Genetic-Proteomic Approach Identifies Residues within Dengue Virus NS4B Critical for Interaction with NS3 and Viral Replication. J. Virol. 2015, 89, 7170–7186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuno, G.; Chang, G.J. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch. Virol. 2007, 152, 687–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baronti, C.; Piorkowski, G.; Charrel, R.N.; Boubis, L.; Leparc-Goffart, I.; de Lamballerie, X. Complete coding sequence of zika virus from a French polynesia outbreak in 2013. Genome Announc. 2014, 2, e00500-14. [Google Scholar] [CrossRef] [PubMed]
- Płaszczyca, A.; Bartenschlager, R. Heidelberg University, Heidelberg, Germany. Replication kinetics of synZIKVs in Huh7 cells. Unpublished work. 2018. [Google Scholar]
- Shan, C.; Xie, X.; Muruato, A.E.; Rossi, S.L.; Roundy, C.M.; Azar, S.R.; Yang, Y.; Tesh, R.B.; Bourne, N.; Barrett, A.D.; et al. An Infectious cDNA Clone of Zika Virus to Study Viral Virulence, Mosquito Transmission, and Antiviral Inhibitors. Cell Host Microbe 2016, 19, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Münster, M.; Bartenschlager, R. Heidelberg University, Heidelberg, Germany. Titration of synZIKV-R2A viruses. Unpublished work. 2017. [Google Scholar]
- Zmurko, J.; Marques, R.E.; Schols, D.; Verbeken, E.; Kaptein, S.J.; Neyts, J. The Viral Polymerase Inhibitor 7-Deaza-2'-C-Methyladenosine Is a Potent Inhibitor of In Vitro Zika Virus Replication and Delays Disease Progression in a Robust Mouse Infection Model. PLoS Negl. Trop. Dis. 2016, 10, e0004695. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Nakayama, Y.; Kinjo, M. Efficient and dynamic nuclear localization of green fluorescent protein via RNA binding. Biochem. Biophys. Res. Commun. 2015, 463, 401–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutso, M.; Saul, S.; Rausalu, K.; Susova, O.; Zusinaite, E.; Mahalingam, S.; Merits, A. Reverse genetic system, genetically stable reporter viruses and packaged subgenomic replicon based on a Brazilian Zika virus isolate. J. Gen. Virol. 2017, 98, 2712–2724. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; Kaufer, B.B. Viral bacterial artificial chromosomes: Generation, mutagenesis, and removal of mini-F sequences. BioMed Res. Int. 2012, 2012, 472537. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shan, C.; Zou, J.; Muruato, A.E.; Bruno, D.N.; de Almeida Medeiros, D.B.; Vasconcelos, P.F.C.; Rossi, S.L.; Weaver, S.C.; Xie, X.; et al. A cDNA Clone-Launched Platform for High-Yield Production of Inactivated Zika Vaccine. EBioMedicine 2017, 17, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Xue, K.S.; Hooper, K.A.; Ollodart, A.R.; Dingens, A.S.; Bloom, J.D. Cooperation between distinct viral variants promotes growth of H3N2 influenza in cell culture. eLife 2016, 5, e13974. [Google Scholar] [CrossRef] [PubMed]
- Scaturro, P.; Trist, I.M.; Paul, D.; Kumar, A.; Acosta, E.G.; Byrd, C.M.; Jordan, R.; Brancale, A.; Bartenschlager, R. Characterization of the mode of action of a potent dengue virus capsid inhibitor. J. Virol. 2014, 88, 11540–11555. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, E.; Brohm, C.; Kallis, S.; Bartenschlager, R.; Pietschmann, T. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J. Virol. 2008, 82, 7034–7046. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Münster, M.; Płaszczyca, A.; Cortese, M.; Neufeldt, C.J.; Goellner, S.; Long, G.; Bartenschlager, R. A Reverse Genetics System for Zika Virus Based on a Simple Molecular Cloning Strategy. Viruses 2018, 10, 368. https://doi.org/10.3390/v10070368
Münster M, Płaszczyca A, Cortese M, Neufeldt CJ, Goellner S, Long G, Bartenschlager R. A Reverse Genetics System for Zika Virus Based on a Simple Molecular Cloning Strategy. Viruses. 2018; 10(7):368. https://doi.org/10.3390/v10070368
Chicago/Turabian StyleMünster, Maximilian, Anna Płaszczyca, Mirko Cortese, Christopher John Neufeldt, Sarah Goellner, Gang Long, and Ralf Bartenschlager. 2018. "A Reverse Genetics System for Zika Virus Based on a Simple Molecular Cloning Strategy" Viruses 10, no. 7: 368. https://doi.org/10.3390/v10070368
APA StyleMünster, M., Płaszczyca, A., Cortese, M., Neufeldt, C. J., Goellner, S., Long, G., & Bartenschlager, R. (2018). A Reverse Genetics System for Zika Virus Based on a Simple Molecular Cloning Strategy. Viruses, 10(7), 368. https://doi.org/10.3390/v10070368