Modulation of the Tumor Microenvironment by CXCR4 Antagonist-Armed Viral Oncotherapy Enhances the Antitumor Efficacy of Dendritic Cell Vaccines against Neuroblastoma in Syngeneic Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Cell Lines
2.2. Viruses
2.3. Generation of Bone Marrow (BM)-Derived DC and In Vitro phagocytosis Assays
2.4. Vaccination of Mice and NXS2 Tumor Challenge
2.5. Treatment of Established Tumors
2.6. Flow Cytometry
2.7. Statistical Analysis
3. Results
3.1. Dox-Induced Apoptosis of NXS2 Tumor Cells is Associated with CRT Exposure and Phagocytosis by DCs
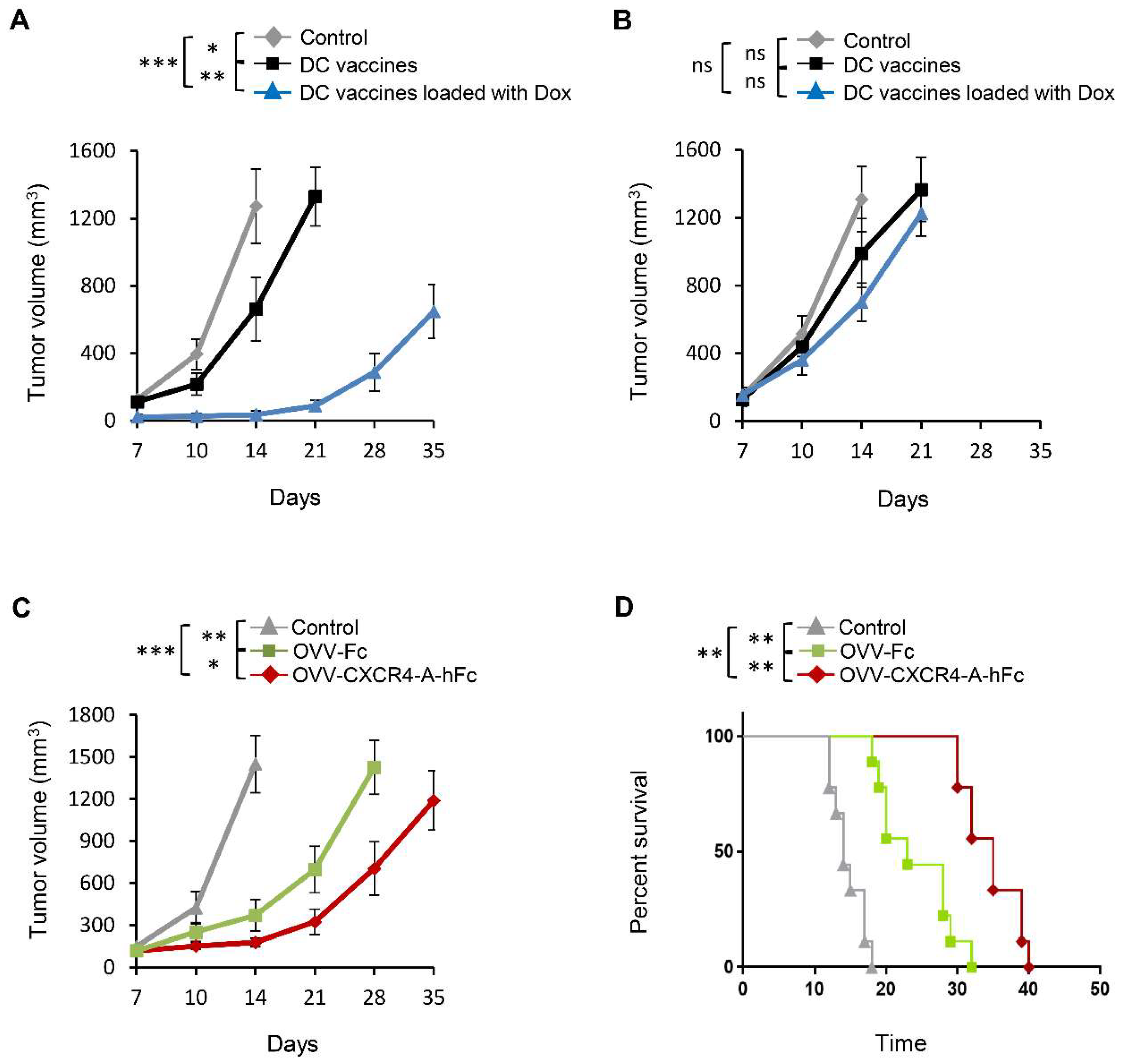
3.2. Vaccination with DC Vaccines Loaded with Dox-Treated NXS2 Tumor Cells Protects Against Syngeneic Tumor Challenge
3.3. OVV-CXCR4-A-Fc Treatment Inhibits Growth of NXS2 Tumors Associated with Decreases in the Immunosuppressive Networks in the TME
3.4. OVV-CXCR4-A-Fc Contributes to Increased Efficacy of the NXS2 Tumor Lysate-Loaded Therapeutic DC Vaccines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Corrias, M.V.; Parodi, S.; Haupt, R.; Lacitignola, L.; Negri, F.; Sementa, A.R.; Dau, D.; Scuderi, F.; Carlini, B.; Bianchi, M.; Casale, F.; et al. Detection of GD2-positive cells in bone marrow samples and survival of patients with localised neuroblastoma. Br. J. Cancer 2008, 98, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, H.V.; Hicks, J.; Okcu, M.F.; Nuchtern, J.G. CXCR4 expression in neuroblastoma primary tumors is associated with clinical presentation of bone and bone marrow metastases. J. Pediatr. Surg. 2004, 39, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yeger, H.; Das, B.; Irwin, M.S.; Baruchel, S. Tissue microenvironment modulates CXCR4 expression and tumor metastasis in neuroblastoma. Neoplasia 2007, 9, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Banisadr, G.; Dicou, E.; Berbar, T.; Rostene, W.; Lombet, A.; Haour, F. Characterization and visualization of [125I] stromal cell-derived factor-1alpha binding to CXCR4 receptors in rat brain and human neuroblastoma cells. J. Neuroimmunol. 2000, 110, 151–160. [Google Scholar] [CrossRef]
- Geminder, H.; Sagi-Assif, O.; Goldberg, L.; Meshel, T.; Rechavi, G.; Witz, I.P.; Ben-Baruch, A. A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J. Immunol. 2001, 167, 4747–4757. [Google Scholar] [CrossRef] [PubMed]
- Libura, J.; Drukala, J.; Majka, M.; Tomescu, O.; Navenot, J.M.; Kucia, M.; Marquez, L.; Peiper, S.C.; Barr, F.G.; Janowska-Wieczorek, A.; et al. CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood 2002, 100, 2597–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuBois, S.G.; Kalika, Y.; Lukens, J.N.; Brodeur, G.M.; Seeger, R.C.; Atkinson, J.B.; Haase, G.M.; Black, C.T.; Perez, C.; Shimada, H.; et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J. Pediatr. Hematol. Oncol. 1999, 21, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Kozin, S.V.; Kirkpatrick, N.D.; Xu, L.; Fukumura, D.; Jain, R.K. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: An emerging sensitizer for anticancer therapies? Clin. Cancer Res. 2011, 17, 2074–2080. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Antiangiogenic agents and targets: A perspective. Biochem. Pharmacol. 2011, 81, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Pol, J.G.; Lichty, B.D.; Cummings, D.T.; Mossman, K.L. Combining oncolytic HSV-1 with immunogenic cell death-inducing drug mitoxantrone breaks cancer immune tolerance and improves therapeutic efficacy. Cancer Immunol. Res. 2013, 1, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Komorowski, M.P.; Seshadri, M.; Rokita, H.; McGray, A.J.; Opyrchal, M.; Odunsi, K.O.; Kozbor, D. CXCL12/CXCR4 Blockade by Oncolytic Virotherapy Inhibits Ovarian Cancer Growth by Decreasing Immunosuppression and Targeting Cancer-Initiating Cells. J. Immunol. 2014, 193, 5327–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, M.; Seshadri, M.; Komorowski, M.P.; Abrams, S.I.; Kozbor, D. Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc. Natl. Acad. Sci. USA 2013, 110, E1291–E1300. [Google Scholar] [CrossRef] [PubMed]
- Permuth-Wey, J.; Lawrenson, K.; Shen, H.C.; Velkova, A.; Tyrer, J.P.; Chen, Z.; Lin, H.Y.; Chen, Y.A.; Tsai, Y.Y.; Qu, X.; et al. Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31. Nat. Commun. 2013, 4, 1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, S.L. Talimogene Laherparepvec: First Global Approval. Drugs 2016, 76, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Fountzilas, C.; Patel, S.; Mahalingam, D. Oncolytic Virotherapy, updates and future directions. Oncotarget 2017, 8, 102617–102639. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.R.; Chow, L.Q.; Nieva, J.; Hwang, T.H.; Moon, A.; Patt, R.; et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011, 477, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.H.; Thorne, S.H. Targeted and armed oncolytic poxviruses: A novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 2009, 9, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.S.; Falls, T.; Burns, J.; Garcia, V.; et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Vanderplasschen, A.; Hollinshead, M.; Smith, G.L. Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and comet formation. J. Gen. Virol. 1997, 78, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Vanderplasschen, A.; Mathew, E.; Hollinshead, M.; Sim, R.B.; Smith, G.L. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc. Natl. Acad. Sci. USA 1998, 95, 7544–7549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Huang, C.T.; Huang, X.; Pardoll, D.M. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat. Immunol. 2004, 5, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Martinez, J.; Huang, X.; Yang, Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood 2007, 109, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Moles, M.A.; Bravo, M.; Ruiz-Avila, I.; Gil-Montoya, J.A.; Acebal, F.; Esteban, F. E-cadherin in non-tumor epithelium adjacent to oral cancer as risk marker for the development of multiple tumors. Br. J. Oral Maxillofac. Surg. 2013, 51, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Sukkurwala, A.; Martins, Q.I.; Wang, Y.; Schlemmer, F.; Ruckenstuhl, C.; Durchschlag, M.; Michaud, M.; Senovilla, L.; Sistigu, A.; Ma, Y.; et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2014, 21, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Reme´dios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Berwin, B.; Hart, J.P.; Rice, S.; Gass, C.; Pizzo, S.V.; Post, S.R.; Nicchitta, C. V. Scavenger receptor-A mediates gp96/GRP94 and calreticulin internalization by antigen-presenting cells. EMBO J. 2003, 22, 6127–6136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berwin, B.; Delneste, Y.; Lovingood, R.V.; Post, S.R.; Pizzo, S.V. SREC-I, a type F scavenger receptor, is an endocytic receptor for calreticulin. J. Biol. Chem. 2004, 279, 51250–51257. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Binder, R.J.; Suto, R.; Anderson, K.M.; Srivastava, P.K. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 2000, 12, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Lode, H.N.; Xiang, R.; Dreier, T.; Varki, N.M.; Gillies, S.D.; Reisfeld, R.A. Natural killer cell-mediated eradication of neuroblastoma metastases to bone marrow by targeted interleukin-2 therapy. Blood 1998, 91, 1706–1715. [Google Scholar] [PubMed]
- Gil, M.; Bieniasz, M.; Wierzbicki, A.; Bambach, B.J.; Rokita, H.; Kozbor, D. Targeting a mimotope vaccine to activating Fcgamma receptors empowers dendritic cells to prime specific CD8+ T cell responses in tumor-bearing mice. J. Immunol. 2009, 183, 6808–6818. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Korz, W. Translating an Antagonist of Chemokine Receptor CXCR4: From bench to bedside. Clin. Cancer Res. 2008, 14, 7975–7980. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yan, W.; Zheng, H.; Du, Q.; Zhang, L.; Ban, Y.; Li, N.; Wei, F. Regulation of IL-10 and IL-12 production and function in macrophages and dendritic cells. F1000 Res. 2015, 4, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.A.; de Cathelineau, A.; Hoffmann, P.R.; Bratton, D.; Ghebrehiwet, B.; Fadok, V.A.; Henson, P.M. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 2001, 194, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Panaretakis, T.; Joza, N.; Tufi, R.; Tesniere, A.; van Endert, P.; Zitvogel, L.; Kroemer, G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007, 14, 1848–1850. [Google Scholar] [CrossRef] [PubMed]
- Cojoc, M.; Peitzsch, C.; Trautmann, F.; Polishchuk, L.; Telegeev, G.D.; Dubrovska, A. Emerging targets in cancer management: Role of the CXCL12/CXCR4 axis. OncoTargets Ther. 2013, 6, 1347–1361. [Google Scholar]
- Gil, M.; Bieniasz, M.; Seshadri, M.; Fisher, D.; Ciesielski, M.J.; Chen, Y.; Pandey, R.K.; Kozbor, D. Photodynamic therapy augments the efficacy of oncolytic vaccinia virus against primary and metastatic tumours in mice. Br. J. Cancer 2011, 105, 1512–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.L.; Benencia, F.; Coukos, G. Whole tumor antigen vaccines. Semin. Immunol. 2010, 22, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, S.; Levy, F.; Burlet-Schiltz, O.; Brasseur, F.; Probst-Kepper, M.; Peitrequin, A.L.; Monsarrat, B.; Van Velthoven, R.; Cerottini, J.C.; Boon, T.; et al. Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity 2000, 12, 107–117. [Google Scholar] [CrossRef]
- Neller, M.A.; Lopez, J.A.; Schmidt, C.W. Antigens for cancer immunotherapy. Semin. Immunol. 2008, 20, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.K. Immunotherapy for human cancer using heat shock protein-peptide complexes. Curr. Oncol. Rep. 2005, 7, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Ridolfi, R.; Riccobon, A.; Galassi, R.; Giorgetti, G.; Petrini, M.; Fiammenghi, L.; Stefanelli, M.; Ridolfi, L.; Moretti, A.; Migliori, G.; et al. Evaluation of in vivo labelled dendritic cell migration in cancer patients. J. Transl. Med. 2004, 2, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilboa, E. DC-based cancer vaccines. J. Clin. Investig. 2007, 117, 1195–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komorowski, M.; Tisonczyk, J.; Kolakowska, A.; Drozdz, R.; Kozbor, D. Modulation of the Tumor Microenvironment by CXCR4 Antagonist-Armed Viral Oncotherapy Enhances the Antitumor Efficacy of Dendritic Cell Vaccines against Neuroblastoma in Syngeneic Mice. Viruses 2018, 10, 455. https://doi.org/10.3390/v10090455
Komorowski M, Tisonczyk J, Kolakowska A, Drozdz R, Kozbor D. Modulation of the Tumor Microenvironment by CXCR4 Antagonist-Armed Viral Oncotherapy Enhances the Antitumor Efficacy of Dendritic Cell Vaccines against Neuroblastoma in Syngeneic Mice. Viruses. 2018; 10(9):455. https://doi.org/10.3390/v10090455
Chicago/Turabian StyleKomorowski, Marcin, Joanna Tisonczyk, Agnieszka Kolakowska, Ryszard Drozdz, and Danuta Kozbor. 2018. "Modulation of the Tumor Microenvironment by CXCR4 Antagonist-Armed Viral Oncotherapy Enhances the Antitumor Efficacy of Dendritic Cell Vaccines against Neuroblastoma in Syngeneic Mice" Viruses 10, no. 9: 455. https://doi.org/10.3390/v10090455