Detection and Characterization of Distinct Alphacoronaviruses in Five Different Bat Species in Denmark

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
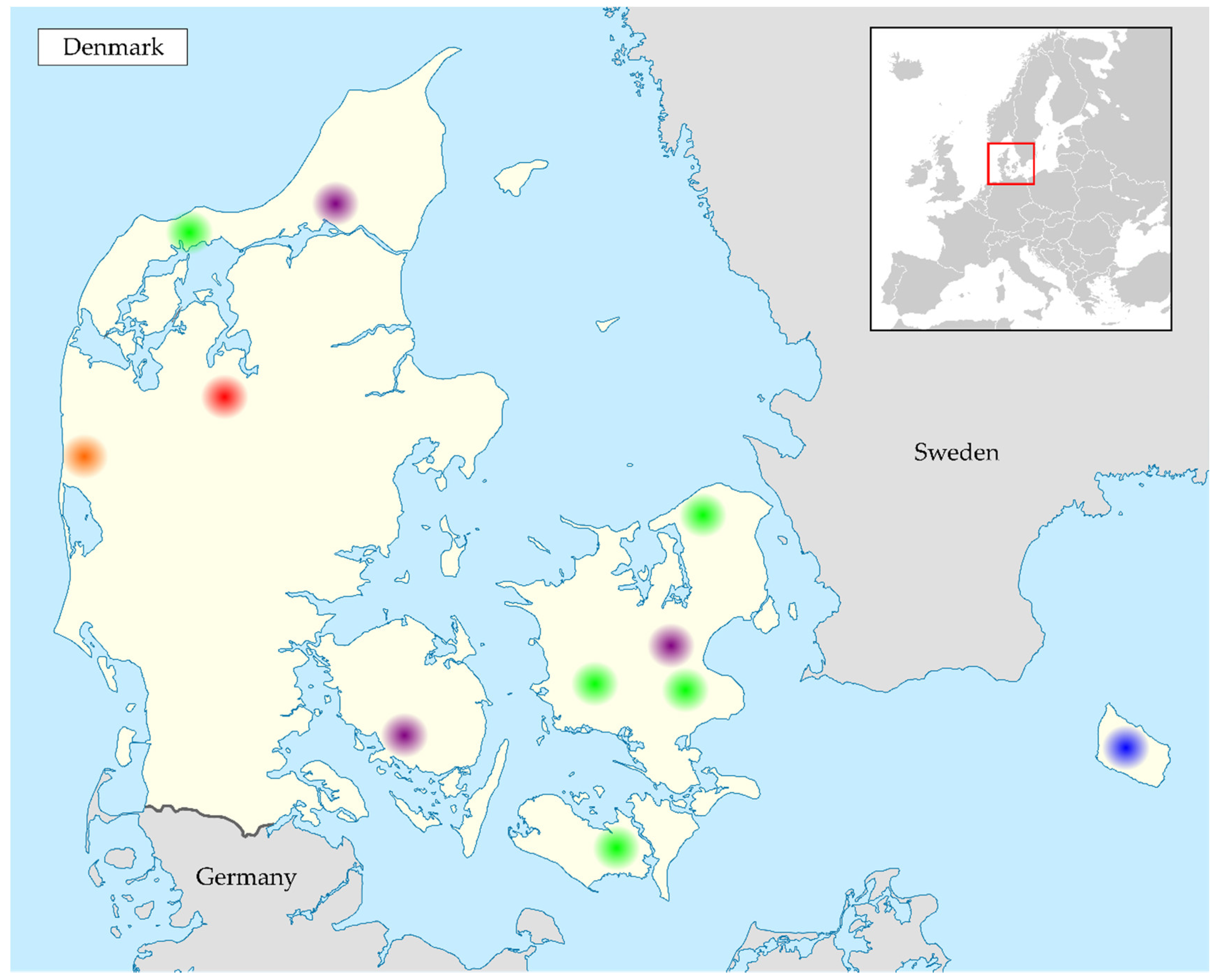
2.1. Collection of Samples
2.2. Screening of Faecal Samples Using pan-CoV RT-PCR Assays
2.3. Sequencing of Samples
2.4. Sequence Analysis
2.5. Statistical Analysis
2.6. Confirmation of Sampled Host Species
3. Results
3.1. Samples Positive for Coronavirus RNA
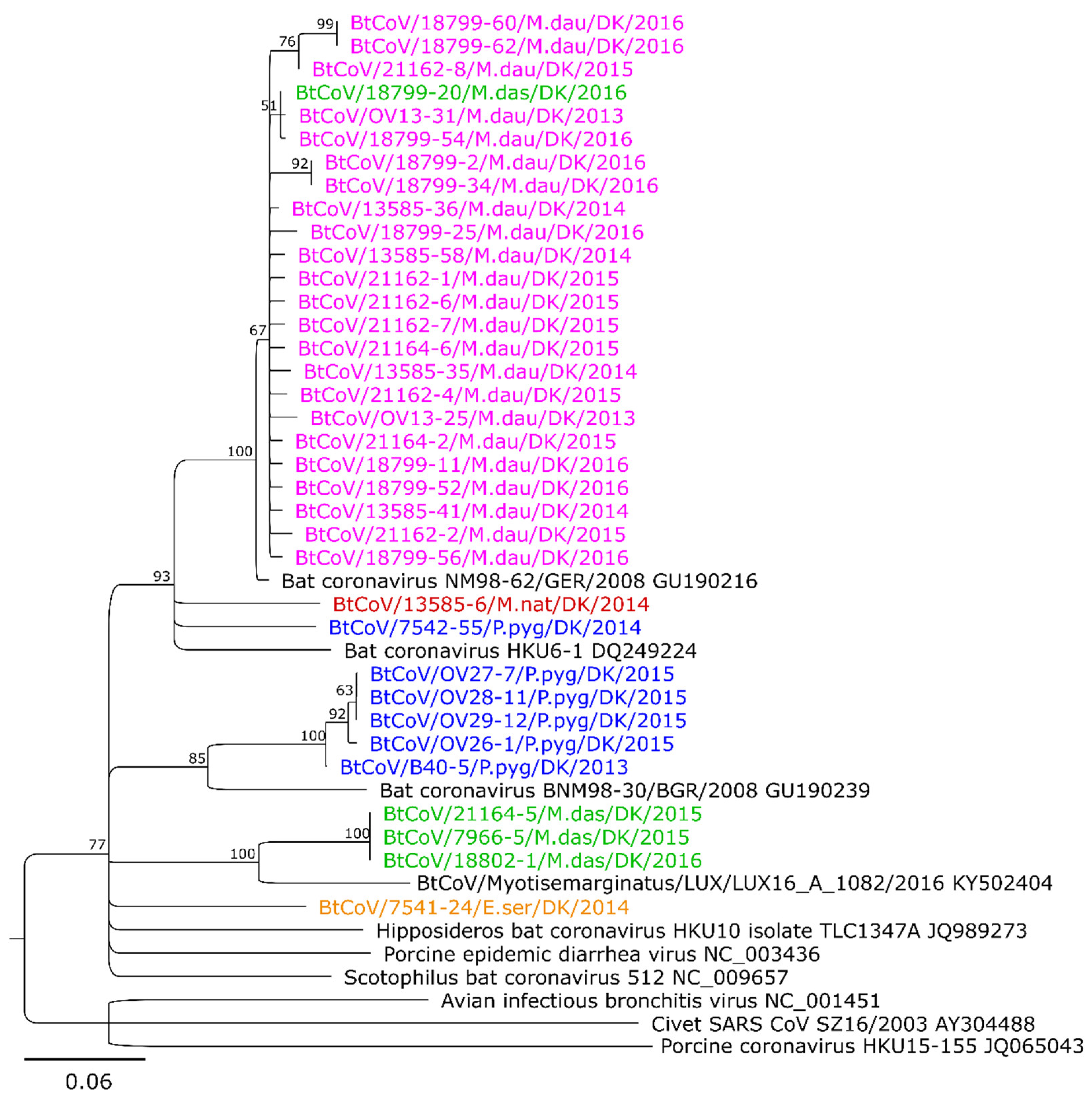
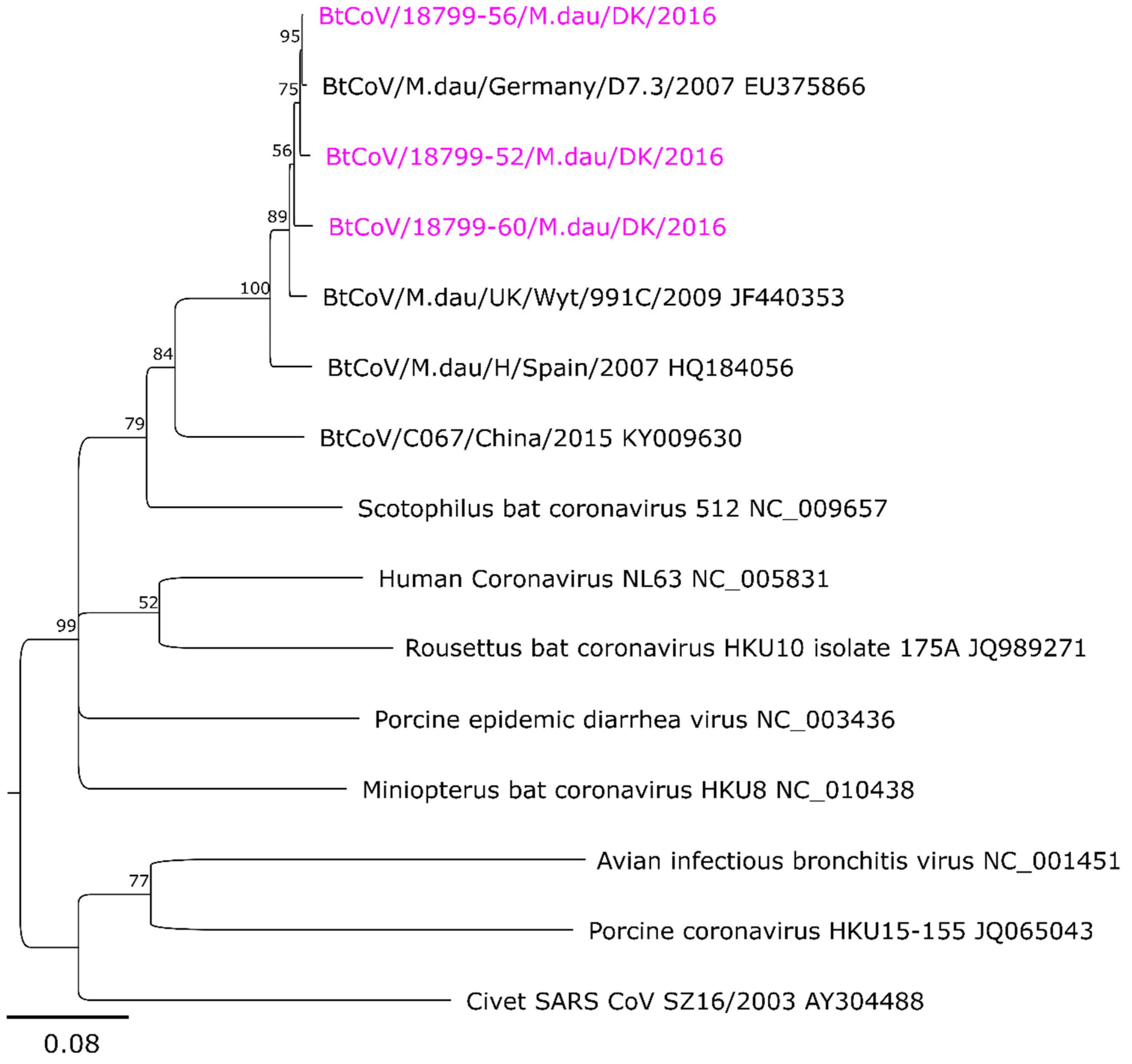
3.2. Sequence Analysis and Phylogeny
3.3. Confirmation of Sampled Host Species
3.4. Predicted Amino Acid Sequences and Host Restriction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavi. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, Evolution and Classification of Bat Coronaviruses in the Aftermath of SARS. Antivir. Res. 2014, 101, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Baagøe, H.J. Danish Bats (Mammalia: Chiroptera): Atlas and Analysis of Distribution, Occurrence and Abundance; Steenstrupia: Copenhagen, Denmark, 2001; ISSN 0375-2909. [Google Scholar]
- Baagøe, H.J. Flagermus, Chiroptera. In Dansk Pattedyratlas; Baagøe, H.J., Jensen, T.S., Eds.; Gyldendal: Copenhagen, Denmark, 2007; pp. 38–98. ISBN 9788702055061. [Google Scholar]
- Baagøe, H.J.; Degn, H.J. Flagermusene i Daugbjerg og Mønsted Kalkgruber i Udflyvningsperioden 2009; Danish Forest and Nature Agency: Midtjylland, Denmark, 2009. [Google Scholar]
- Møller, J.D.; Baagøe, H.J.; Degn, H.J. Forvaltningsplan for Flagermus; Naturstyrelsen, Miljøministeriet: Copenhagen, Denmark, 2013; ISBN 9788772794075. [Google Scholar]
- Constantine, D.G. An Automatic Bat-Collecting Device. J. Wildl. Manag. 1958, 22, 17–22. [Google Scholar] [CrossRef]
- Tuttle, M.D. An Improved Trap for Bats. J. Mammal. 1974, 55, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Baagøe, H.J.; Degn, H.J. Flagermusene i Daugbjerg og Mønsted Kalkgruber i Udflyvningsperioden 2003; National Environmental Research Institute of Denmark: Roskilde, Denmark, 2004. [Google Scholar]
- Escutenaire, S.; Mohamed, N.; Isaksson, M.; Thorén, P.; Klingeborn, B.; Belák, S.; Berg, M.; Blomberg, J. SYBR Green Real-Time Reverse Transcription-Polymerase Chain Reaction Assay for the Generic Detection of Coronaviruses. Arch. Virol. 2007, 152, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Moës, E.; Keyaerts, E.; Li, S.; Van Ranst, M. A Pancoronavirus RT-PCR Assay for Detection of All Known Coronaviruses. Methods Mol. Biol. 2008, 454, 3–12. [Google Scholar] [CrossRef] [PubMed]
- De Souza Luna, L.K.; Heiser, V.; Regamey, N.; Panning, M.; Drexler, J.F.; Mulangu, S.; Poon, L.; Baumgarte, S.; Haijema, B.J.; Kaiser, L.; et al. Generic Detection of Coronaviruses and Differentiation at the Prototype Strain Level by Reverse Transcription-PCR and Nonfluorescent Low-Density Microarray. J. Clin. Microbiol. 2007, 45, 1049–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Committee on Taxonomy of Viruses (ICTV) Virus Metadata Repository: Version 21 March 2018; MSL32. Available online: https://talk.ictvonline.org/taxonomy/vmr/ (accessed on 15 May 2018).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Aragon, T.J. Epitools: Epidemiology Tools. Available online: https://cran.r-project.org/package=epitools (accessed on 15 May 2018).
- Schlegel, M.; Ali, H.S.; Stieger, N.; Groschup, M.H.; Wolf, R.; Ulrich, R.G. Molecular Identification of Small Mammal Species Using Novel Cytochrome b Gene-Derived Degenerated Primers. Biochem. Genet. 2012, 50, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic Characterization of Severe Acute Respiratory Syndrome-Related Coronavirus in European Bats and Classification of Coronaviruses Based on Partial RNA-Dependent RNA Polymerase Gene Sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Göttsche, M.; Panning, M.; Drexler, J.F.; Petersen, N.; Annan, A.; Grywna, K.; Müller, M.; et al. Detection and Prevalence Patterns of Group I Coronaviruses in Bats, Northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Falcón, A.; Vázquez-Morón, S.; Casas, I.; Aznar, C.; Ruiz, G.; Pozo, F.; Perez-Breña, P.; Juste, J.; Ibáñez, C.; Garin, I.; et al. Detection of Alpha and Betacoronaviruses in Multiple Iberian Bat Species. Arch. Virol. 2011, 156, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- August, T.A.; Mathews, F.; Nunn, M.A. Alphacoronavirus Detected in Bats in the United Kingdom. Vector-Borne Zoonotic Dis. 2012, 12, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Goffard, A.; Demanche, C.; Arthur, L.; Pinçon, C.; Michaux, J.; Dubuisson, J. Alphacoronaviruses detected in french bats are phylogeographically linked to coronaviruses of european bats. Viruses 2015, 7, 6279–6290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemenesi, G.; Dallos, B.; Gorfol, T.; Boldogh, S.; Estok, P.; Kurucz, K.; Kutas, A.; Foldes, F.; Oldal, M.; Nemeth, V.; et al. Molecular survey of RNA viruses in hungarian bats: Discovering novel astroviruses, coronaviruses, and caliciviruses. Vector-Borne Zoonotic Dis. 2014, 14, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Müller, M.A.; Drosten, C. Amplification of Emerging Viruses in a Bat Colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.; Edenborough, K.M.; Toffoli, R.; Culasso, P.; Zoppi, S.; Dondo, A.; Robetto, S.; Rosati, S.; Lander, A.; Kurth, A.; et al. Coronavirus and Paramyxovirus in Bats from Northwest Italy. BMC Vet. Res. 2017, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Smith, C.S.; Peel, A.J.; Plowright, R.K.; Kerlin, D.H.; McBroom, J.; McCallum, H. Persistent Infections Support Maintenance of a Coronavirus in a Population of Australian Bats (Myotis macropus). Epidemiol. Infect. 2017, 145, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Rasche, A.; Diallo, T.D.; Cottontail, V.M.; Stöcker, A.; Souza, B.F.d.C.D.; Corrêa, J.I.; Carneiro, A.J.B.; Franke, C.R.; Nagy, M.; et al. Highly Diversified Coronaviruses in Neotropical Bats. J. Gen. Virol. 2013, 94, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Han, N.; Streicker, D.; Li, G.; Tang, X.; Shi, Z.; Hu, Z.; Zhao, G.; Fontanet, A.; Guan, Y.; et al. Evolutionary Relationships between Bat Coronaviruses and Their Hosts. Emerg. Infect. Dis. 2007, 13, 1526–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.C.; Zhang, J.X.; Zhang, S.Y.; Wang, P.; Fan, X.H.; Li, L.F.; Li, G.; Dong, B.Q.; Liu, W.; Cheung, C.L.; et al. Prevalence and Genetic Diversity of Coronaviruses in Bats from China. J. Virol. 2006, 80, 7481–7490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Liu, Y.; Weiss, S.; Arnold, E.; Sarafianos, S.G.; Ding, J. Molecular Model of SARS Coronavirus Polymerase: Implications for Biochemical Functions and Drug Design. Nucleic Acids Res. 2003, 31, 7117–7130. [Google Scholar] [CrossRef] [PubMed]
- Gaisler, J.; Hanak, V.; Hanzal, V.; Jarský, V. Results of bat banding in the Czech and Slovak Republics, 1948–2000. Vespertilio 2003, 7, 3–61. [Google Scholar]
- Dietz, C.; von Helversen, O.; Nill, D. Bats of Britain, Europe and Northwest Africa, 1st ed.; A & C Black Publishers Ltd.: London, UK, 2009; ISBN 9781408105313. [Google Scholar]
- Steffens, R.; Zöphel, U.; Brockmann, D. 40th Anniversary Bat Marking Centre Dresden—Evaluation of Methods and Overview of Results; Saxon State Office for Environment and Geology: Dresden, Germany, 2004; ISBN 3-00-016143-0. [Google Scholar]
- Ahlén, I.; Baagøe, H.J.; Bach, L. Behavior of Scandinavian Bats during Migration and Foraging at Sea. J. Mammal. 2009, 90, 1318–1323. [Google Scholar] [CrossRef] [Green Version]
- Egsbæk, W.; Jensen, B. Results of bat banding in Denmark. Vidensk. Medd. Dansk Naturh. Foren. 1963, 125, 269–296. [Google Scholar]
- Baagøe, H.J. Bornholms flagermus-status 2010. Nat. Bornh. 2011, 9, 22–30. [Google Scholar]
- Borkenhagen, P. Die Säugetiere Schleswig-Holsteins, 1st ed.; Husum Druck- und Verlagsgesellschaft: Husum, Germany, 2011; ISBN 978-3898765619. [Google Scholar]
- Reusken, C.C.B.E.M.; Lina, P.H.C.C.; Pielaat, A.; de Vries, A.; Dam-Deisz, C.; Adema, J.; Drexler, J.F.; Drosten, C.; Kooi, E.A. Circulation of Group 2 Coronaviruses in a Bat Species Common to Urban Areas in Western Europe. Vector-Borne Zoonotic Dis. 2010, 10, 785–791. [Google Scholar] [CrossRef] [PubMed]
- De Benedictis, P.; Marciano, S.; Scaravelli, D.; Priori, P.; Zecchin, B.; Capua, I.; Monne, I.; Cattoli, G. Alpha and Lineage C BetaCoV Infections in Italian Bats. Virus Genes 2014, 48, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Annan, A.; Baldwin, H.J.; Corman, V.M.; Klose, S.M.; Owusu, M.; Nkrumah, E.E.; Badu, E.K.; Anti, P.; Agbenyega, O.; Meyer, B.; et al. Human Betacoronavirus 2c EMC/2012-Related Viruses in Bats, Ghana and Europe. Emerg. Infect. Dis. 2013, 19, 456–459. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bat Species | No. Sampled (No. Positive) | Collection Date | Collection Location | Sample Types |
|---|---|---|---|---|
| M. daubentonii | 4 (0) | September 2013 | Vesløs | 2 individuals and 2 from nest box |
| 40 (8) | October 2013 | Mønsted | Individual | |
| 53 (9) | October 2014 | Mønsted | Individual | |
| 4 (1) | April 2015 | Mønsted | Individual | |
| 10 (6) | October 2015 | Mønsted | Individual | |
| 11 (6) | November 2015 | Mønsted | Individual | |
| 5 (0) | May 2016 | Mønsted | Individual | |
| 58 (13) | September 2016 | Mønsted | Individual | |
| 2 (0) | April 2017 | Mønsted | Individual | |
| M. dasycneme | 2 (0) | October 2013 | Mønsted | Individual |
| 6 (0) | October 2014 | Mønsted | Individual | |
| 2 (1) | April 2015 | Mønsted | Individual | |
| 1 (1) | November 2015 | Mønsted | Individual | |
| 6 (1) | September 2016 | Mønsted | Individual | |
| 2 (2) | November 2016 | Mønsted | Individual | |
| M. nattereri | 7 (0) | August–September 2013 | Bornholm | Individual |
| 1 (0) | September 2013 | Lolland, Agerup forest | Individual | |
| 2 (1) | October 2014 | Mønsted | Individual | |
| 1 (0) | September 2016 | Mønsted | Individual | |
| M. bechsteinii | 4 (0) | August 2013 | Bornholm | Individual |
| 2 (0) | August 2015 | Bornholm | 1 individual and 1 from nest box | |
| 3 (0) | August 2016 | Bornholm | 3 nest boxes | |
| 2 (0) | September 2017 | Bornholm | Individual | |
| M. mystacinus | 1 (0) | August 2013 | Bornholm | Individual |
| 1 (0) | August 2016 | Bornholm | Individual | |
| M. brandtii | 2 (0) | August 2013 | Bornholm | Individual |
| 1 (0) | April 2015 | Mønsted | Individual | |
| N. noctula | 4 (0) | August 2013 | Bornholm | 2 individual samples and 2 from nest boxes |
| P. pygmaeus | 6 (1) | August 2013 | Funen, Sollerup forest | Individual |
| 9 (1) | June 2014 | Vadum | Mix of individual samples and floor samples from colony | |
| 1 (0) | Summer 2015 | Sorø | Sample from nest box | |
| 8 (4) | September 2015 | Borup | 5 individual samples, 3 from floor | |
| 1 (0) | September–October 2015 | Helsinge | Sample from abandoned colony | |
| 3 (0) | August 2016 | Tureby | From colony | |
| P. auritus | 1 (0) | August 2013 | Bornholm | Individual |
| 1 (0) | September–October 2015 | Bornholm | Sample from floor | |
| E. serotinus | 3 (3) | June 2014 | Tim | 1 individual sample, 2 from floor |
| 1 (0) | June 2017 | Sakskøbing | Sample from floor |
| Bat Species | Total No. Sampled (No. Positive) | Observed Coronavirus Prevalence in Samples | 95% Confidence Interval of Sample Mean |
|---|---|---|---|
| M. daubentonii | 187 (43) | 23% | [0.17; 0.29] |
| M. dasycneme | 19 (5) | 26% | [0.07; 0.46] |
| M. nattereri | 11 (1) | 9% | [0.00; 0.26] |
| M. bechsteinii | 11 (0) | 0% | NA 1 |
| M. mystacinus | 2 (0) | 0% | NA |
| M. brandtii | 3 (0) | 0% | NA |
| N. noctula | 4 (0) | 0% | NA |
| P. pygmaeus | 28 (6) | 21% | [0.06; 0.37] |
| P. auritus | 2 (0) | 0% | NA |
| E. serotinus | 4 (3) | 75% | [0.33; 1.00] |
| Total (all species) | 271 (58) | 21% | [0.17; 0.26] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazov, C.M.; Chriél, M.; Baagøe, H.J.; Fjederholt, E.; Deng, Y.; Kooi, E.A.; Belsham, G.J.; Bøtner, A.; Rasmussen, T.B. Detection and Characterization of Distinct Alphacoronaviruses in Five Different Bat Species in Denmark. Viruses 2018, 10, 486. https://doi.org/10.3390/v10090486
Lazov CM, Chriél M, Baagøe HJ, Fjederholt E, Deng Y, Kooi EA, Belsham GJ, Bøtner A, Rasmussen TB. Detection and Characterization of Distinct Alphacoronaviruses in Five Different Bat Species in Denmark. Viruses. 2018; 10(9):486. https://doi.org/10.3390/v10090486
Chicago/Turabian StyleLazov, Christina M., Mariann Chriél, Hans J. Baagøe, Esben Fjederholt, Yu Deng, Engbert A. Kooi, Graham J. Belsham, Anette Bøtner, and Thomas Bruun Rasmussen. 2018. "Detection and Characterization of Distinct Alphacoronaviruses in Five Different Bat Species in Denmark" Viruses 10, no. 9: 486. https://doi.org/10.3390/v10090486
APA StyleLazov, C. M., Chriél, M., Baagøe, H. J., Fjederholt, E., Deng, Y., Kooi, E. A., Belsham, G. J., Bøtner, A., & Rasmussen, T. B. (2018). Detection and Characterization of Distinct Alphacoronaviruses in Five Different Bat Species in Denmark. Viruses, 10(9), 486. https://doi.org/10.3390/v10090486