Co-Circulation and Excretion Dynamics of Diverse Rubula- and Related Viruses in Egyptian Rousette Bats from South Africa

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
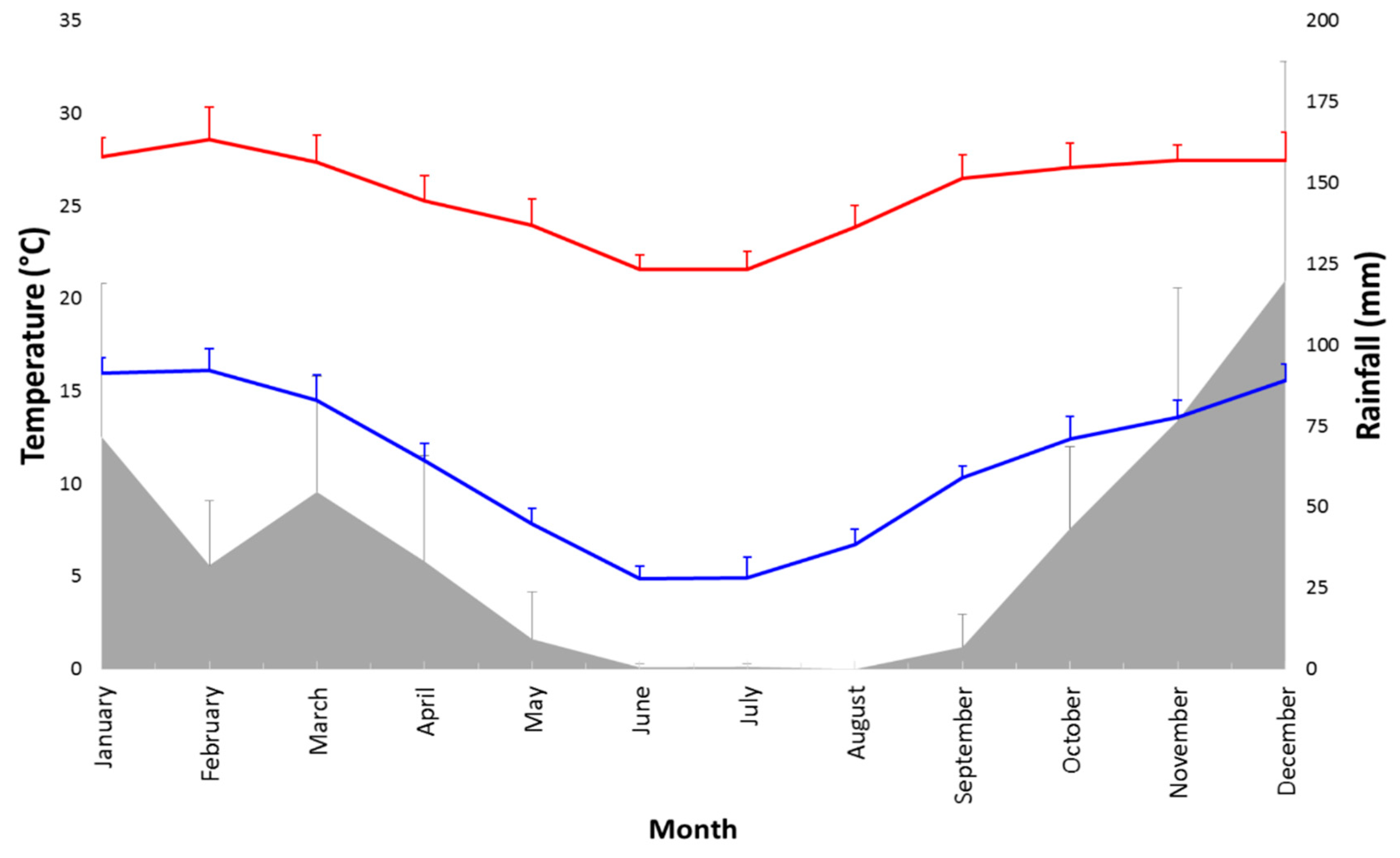
2.1. Study Site, Permits, and Ethical Statements
2.2. Sample Collection
2.3. Viral PCR Screening
2.4. Bioinformatic Analysis
2.5. Temporal Analysis
3. Results
3.1. Positivity and Co-Infections
3.2. Phylogenetic Analysis and Identity Comparisons
3.3. Tissue Distribution
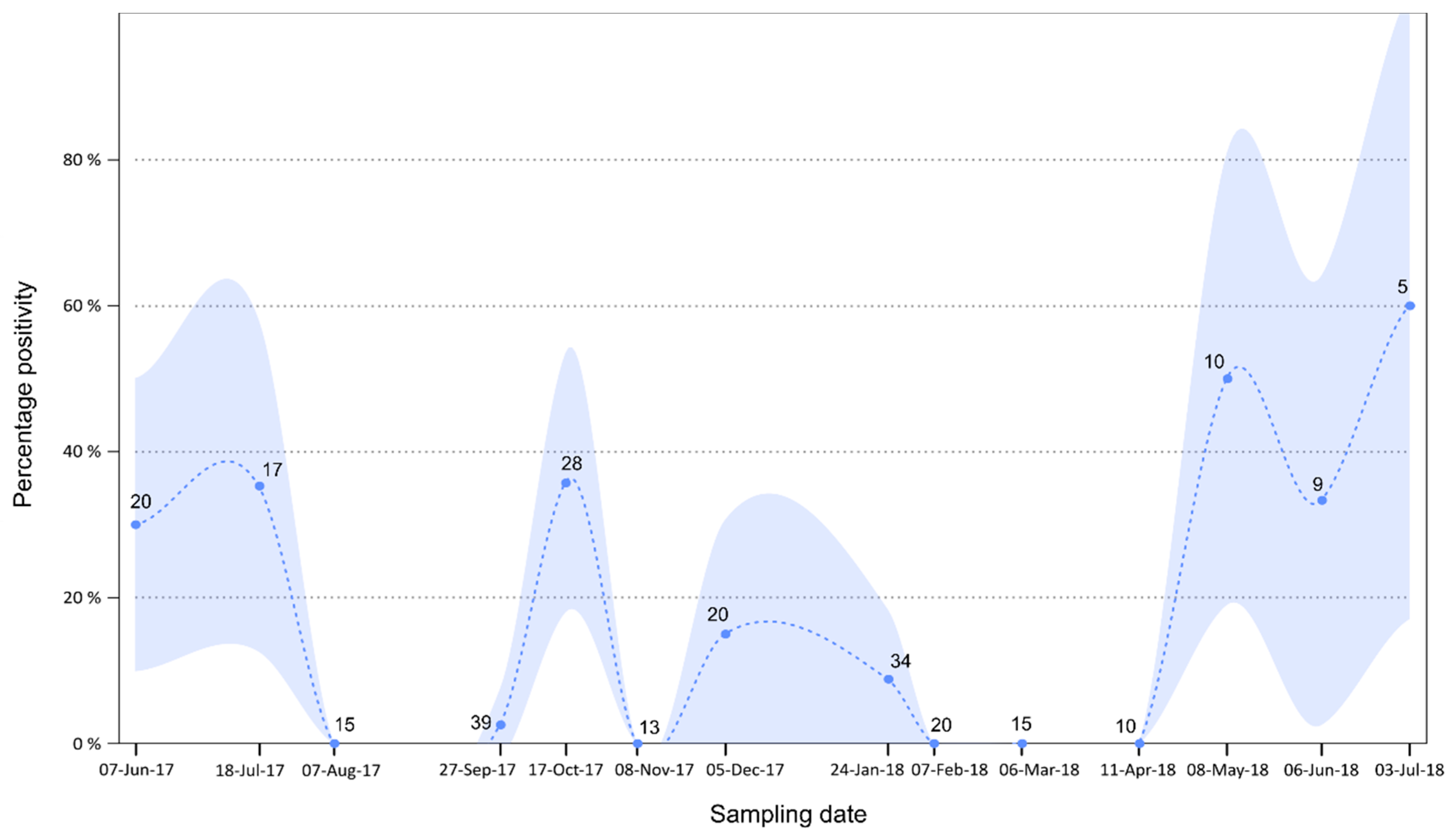
3.4. Temporal Analysis
4. Discussion
4.1. Detection of Nucleic Acids
4.2. Tissue Distribution
4.3. Excretion Dynamics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



References
- Luis, A.D.; Hayman, D.T.S.; O’Shea, T.J.; Cryan, P.M.; Gilbert, A.T.; Pulliam, J.R.C.; Mills, J.N.; Timonin, M.E.; Willis, C.K.R.; Cunningham, A.A.; et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proc. R. Soc. B Biol. Sci. 2013, 280. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, G.K.; Aréchiga Ceballos, N.G.; Banyard, A.C.; Basler, C.F.; Bavari, S.; Bennett, A.J.; Blasdell, K.R.; Briese, T.; Bukreyev, A.; Caì, Y.; et al. Taxonomy of the order Mononegavirales: Update 2017. Arch. Virol. 2017, 162, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Selleck, P.; Hooper, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Westbury, H.; Hiley, L.; Selvey, L.; Rodwell, B.; et al. A morbillivirus that caused fatal disease in horses and humans. Science 1995, 268, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Halpin, K.; Young, P.L.; Field, H.E.; Mackenzie, J.S. Isolation of Hendra virus from pteropid bats: A natural reservoir of Hendra virus. J. Gen. Virol. 2000, 81, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Shieh, W.; Goldsmith, C.S.; Gubler, D.J.; Roehrig, J.T.; Ling, A.E.; Peters, C.J.; et al. Nipah virus: A recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Paul, L. Nipah virus in Kerala: A deadly zoonosis. Clin. Microbiol. Infect. 2018, 24, 1113–1114. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Rasche, A.; Yordanov, S.; Seebens, A.; et al. Bats host major mammalian paramyxoviruses. PLoS ONE 2012, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.S.; Todd, S.; Marsh, G.; Fernandez-Loras, A.; Suu-Ire, R.; Wood, J.L.N.; Wang, L.F.; Murcia, P.R.; Cunningham, A.A. Co-circulation of diverse paramyxoviruses in an urban African fruit bat population. J. Gen. Virol. 2012, 93, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Muleya, W.; Sasaki, M.; Orba, Y.; Ishii, A.; Thomas, Y.; Nakagawa, E.; Ogawa, H.; Hang’ombe, B.; Namangala, B.; Mweene, A.; et al. Molecular Epidemiology of Paramyxoviruses in Frugivorous Eidolon helvum Bats in Zambia. J. Vet. Med. Sci. 2014, 76, 611–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortlock, M.; Kuzmin, I.V.; Weyer, J.; Gilbert, A.T.; Agwanda, B.; Rupprecht, C.E.; Nel, L.H.; Kearney, T.; Malekani, J.M.; Markotter, W. Novel paramyxoviruses in bats from Sub-Saharan Africa, 2007–2012. Emerg. Infect. Dis. 2015, 21, 1840–1843. [Google Scholar] [CrossRef]
- Virtue, E.R.; Marsh, G.A.; Wang, L. Paramyxoviruses infecting humans: The old, the new and the unknown. Future Microbiol. 2009, 4, 537–554. [Google Scholar] [CrossRef] [PubMed]
- Hagmaier, K.; Stock, N.; Precious, B.; Childs, K.; Wang, L.; Goodbourn, S.; Randall, R.E. Mapuera virus, a rubulavirus that inhibits interferon signalling in a wide variety of mammalian cells without degrading STATs. J. Gen. Virol. 2007, 88, 956–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, A.; Zhu, Z.; Hu, Y.; Deng, X.; Sun, Z.; Zhang, Y.; Mao, N.; Xu, S.; Fang, X.; Gao, H.; et al. Mumps epidemiology and mumps virus genotypes circulating in mainland China during 2013–2015. PLoS ONE 2017, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schaap-Nutt, A.; Higgins, C.; Amaro-Carambot, E.; Nolan, S.M.; D’Angelo, C.; Murphy, B.R.; Collins, P.L.; Schmidt, A.C. Identification of human parainfluenza virus type 2 (HPIV-2) V protein amino acid residues that reduce binding of V to MDA5 and attenuate HPIV-2 replication in nonhuman primates. J. Virol. 2011, 85, 4007–4019. [Google Scholar] [CrossRef] [PubMed]
- Chant, K.; Chan, R.; Smith, M.; Dwyer, D.E.; Kirkland, P.; Conaty, S.; Cossart, Y.; Gilbert, G.; Jane, R.; Love, R.; et al. Probable human infection with a newly described virus in the family Paramyxoviridae. Emerg. Infect. Dis. 1998, 4, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.A.; Smith, C.; Marsh, G.A.; Field, H.; Wang, L. Evidence of bat origin for Menangle virus, a zoonotic paramyxovirus first isolated from diseased pigs. J. Gen. Virol. 2012, 93, 2590–2594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albariño, C.G.; Foltzer, M.; Towner, J.S.; Rowe, L.A.; Campbell, S.; Jaramillo, C.M.; Bird, B.H.; Reeder, D.M.; Vodzak, M.E.; Rota, P.; et al. Novel paramyxovirus associated with severe acute febrile disease, South Sudan and Uganda, 2012. Emerg. Infect. Dis. 2014, 20, 211–216. [Google Scholar] [CrossRef]
- Amman, B.R.; Albariño, C.G.; Bird, B.H.; Nyakarahuka, L.; Sealy, T.K.; Balinandi, S.; Schuh, A.J.; Campbell, S.M.; Ströher, U.; Megan, E.B.; et al. A recently discovered pathogenic paramyxovirus, Sosuga Virus, is present in Rousettus aegyptiacus fruit bats at multiple locations in Uganda. J. Wildl. Dis. 2015, 51, 774–779. [Google Scholar] [CrossRef]
- Barr, J.A.; Todd, S.; Crameri, G.; Foord, A.; Marsh, G.; Frazer, L.; Payne, J.; Harper, J.; Baker, K.S.; Cunningham, A.A.; et al. Animal infection studies of two recently discovered African bat paramyxoviruses, Achimota 1 and Achimota 2. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Katoh, H.; Kubota, T.; Ihara, T.; Maeda, K.; Takeda, M.; Kidokoro, M. Cross-neutralization between human and African bat mumps viruses. Emerg. Infect. Dis. 2016, 22, 703–706. [Google Scholar] [CrossRef]
- Krüger, N.; Hoffmann, M.; Drexler, J.F.; Müller, M.A.; Corman, V.M.; Sauder, C.; Rubin, S.; He, B.; Örvell, C.; Drosten, C.; Herrler, G. Functional properties and genetic relatedness of the fusion and hemagglutinin-neuraminidase proteins of a mumps virus-like bat virus. J. Virol. 2015, 89, 4539–4548. [Google Scholar] [CrossRef] [PubMed]
- Beaty, S.M.; Nachbagauer, R.; Hirsh, A.; Vigant, F.; Duehr, J.; Azarm, K.D.; Stelfox, A.J.; Bowden, T.A.; Duprex, W.P.; Krammer, F.; et al. Cross-reactive and cross-neutralizing activity of human mumps antibodies against a novel mumps virus from bats. J. Infect. Dis. 2017, 215, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinski, G.G.; Griffiths, T.A. Rousettus aegyptiacus. Mamm. Species 1999, 611, 1–9. [Google Scholar] [CrossRef]
- Towner, J.S.; Pourrut, X.; Albariño, C.G.; Nkogue, C.N.; Bird, B.H.; Grard, G.; Ksiazek, T.G.; Gonzalez, J.P.; Nichol, S.T.; Leroy, E.M. Marburg virus infection detected in a common African bat. PLoS ONE 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Swanepoel, R.; Smit, S.B.; Rollin, P.E.; Formenty, P.; Leman, P.A.; Kemp, A.; Burt, F.J.; Grobbelaar, A.A.; Croft, J.; Bausch, D.G.; et al. Studies of Reservoir Hosts for Marburg Virus. Emerg. Infect. Dis. 2007, 13, 1847–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paweska, J.T.; Jansen van Vuren, P.; Kemp, A.; Storm, N.; Grobbelaar, A.A.; Wiley, M.R.; Palacios, G.; Markotter, W. Marburg virus infection in Egyptian rousette bats, South Africa, 2013–2014. Emerg. Infect. Dis. 2018, 24, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, I.V.; Niezgoda, M.; Franka, R.; Agwanda, B.; Markotter, W.; Beagley, J.C.; Urazova, O.Y.; Breiman, R.F.; Rupprecht, C.E. Lagos bat virus in Kenya. J. Clin. Microbiol. 2008, 46, 1451–1461. [Google Scholar] [CrossRef]
- Jacobsen, N.H.G.; Du Plessis, E. Observations on the ecology and biology of the Cape fruit bat Rousettus aegyptiacus leachi in the Eastern Transvaal. S. Afr. J. Sci. 1976, 72, 270–273. [Google Scholar]
- Tong, S.; Chern, S.-W.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Amarasinghe, G.K.; Aréchiga Ceballos, N.G.; Banyard, A.C.; Basler, C.F.; Bavari, S.; Bennett, A.J.; Blasdell, K.R.; Briese, T.; Bukreyev, A.; Caì, Y.; et al. Taxonomy of the order Mononegavirales: Update 2018. Arch. Virol. 2018, 163, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. Int. Soc. Microb. Ecol. J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Tsurudome, M.; Bando, H.; Ito, Y. Immunological relationships of simian virus 41 (SV41) to other paramyxoviruses and serological evidence of SV41 infection in human populations. J. Gen. Virol. 1990, 71, 2093–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapanes, E.; Detwiler, K.M.; Cords, M. Bat predation by cercopithecus monkeys: Implications for zoonotic disease transmission. Ecohealth 2016, 13, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Mickleburgh, S.; Waylen, K.; Racey, P. Bats as bushmeat: A global review. Oryx 2009, 43, 217–234. [Google Scholar] [CrossRef]
- Pernet, O.; Schneider, B.S.; Beaty, S.M.; LeBreton, M.; Yun, T.E.; Park, A.; Zachariah, T.T.; Bowden, T.A.; Hitchens, P.; Ramirez, C.M.; et al. Evidence for henipavirus spillover into human populations in Africa. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Chua, K.B.; Koh, C.L.; Hooi, P.S.; Wee, K.F.; Khong, J.H.; Chua, B.H.; Chan, Y.P.; Lim, M.E.; Lam, S.K. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 2002, 4, 145–151. [Google Scholar] [CrossRef]
- Plowright, R.K.; Field, H.E.; Smith, C.; Divljan, A.; Palmer, C.; Tabor, G.; Daszak, P.; Foley, J.E. Reproduction and nutritional stress are risk factors for Hendra virus infection in little red flying foxes (Pteropus scapulatus). Proc. R. Soc. B Biol. Sci. 2008, 275, 861–869. [Google Scholar] [CrossRef]
- Dietrich, M.; Wilkinson, D.A.; Benlali, A.; Lagadec, E.; Ramasindrazana, B.; Dellagi, K.; Tortosa, P. Leptospira and paramyxovirus infection dynamics in a bat maternity enlightens pathogen maintenance in wildlife. Environ. Microbiol. 2015, 17, 4280–4289. [Google Scholar] [CrossRef]
- Mutere, F.A. The breeding biology of the fruit bat Rousettus aegyptiacus E. Geoffroy living at 0°22′S. Acta Trop. 1968, 25, 97–108. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Representative Virus Sequence † (Number of Detections) | Samples Positive (Sample Type) *,^ | GenBank Accession Numbers | Highest Similarity (%) to Classified Rubulavirus Species # | |||
|---|---|---|---|---|---|---|
| Virus | nt | Virus | aa | |||
| BatPV_R_aeg_RSA-1497_2012 (11) | UP 1497 (S), UP 3584 (S), UP 4251 (S), UP 4260 (S), UP 4488 (U), UP 5261 (U),UPE 091 (U), UPE 326 (U), UPE 337 (U), UPE 479 (U), UPE 489 (U) | MH259215; MH259227; MH259233; MH259235; MH259263; MH259264; MH259270; MH259289; MH259292; MH259296; MH259298 | AchPV-1 | 76.3% | AchPV-1, AchPV-2, ThkPV-1, ThkPV-2, Sosuga | 82.2% |
| BatPV_R_aeg_RSA-1501_2012 (4) | UP 1501 (S) UP 2729 (S), UP 5862 (S),UPE 094 (U) | MH259216; MH259221; MH259240; MH259272 | ThkPV-1 | 78.4% | ThkPV-1 | 83.8% |
| BatPV_R_aeg_RSA-1511_2012 (2) | UP 1511 (S), UPE 816 (U) | MH259217; MH937577 | AchPV-2 | 76.3% | AchPV-2 | 83.8% |
| BatPV_R_aeg_RSA-1519_2012 (3) | UP 1519 (S), UP 3011 (S), UP 4729 (S) | MH259218; MH259225; MH259238 | ThkPV-1 | 72% | AchPV-2, TioPV | 80.6% |
| BatPV_R_aeg_RSA-2049_2012 (24) | UP 2049 (S), UP 2240 (U), UP 3093 (S), UP 3760 (S), UP 5119 (S), UP 5908 (S), UP 6101 (S), UP 6892 (S), UP 6910 (S), UPE 088 (U), UPE 113 (U), UPE 117 (U), UPE 195 (U), UPE 337 (U), UPE 527 (U), UPE 529 (U), UPE 761 (U), UPE 762 (U), UPE 764 (U), UPE 766 (U), UPE 769 (U), UPE 789 (U), UPE 808 (U), UPE 813 (U) | MH259219; MH259262; MH259226; MH259229; MH259239; MH259241; MH259242; MH259245; MH259265; MH259269; MH259274; MH259278; MH259284; MH259293; MH259300; MH259301; MH937567; MH937568; MH937569; MH937570; MH937571; MH937572; MH937573; MH937575 | MenPV | 74.1% | AchPV-2 | 82.2% |
| BatPV_R_aeg_RSA-2240a_2013 (1) | UP 2240 (U) | MH259260 | ThkPV-2 | 75.8% | Sosuga | 79% |
| BatPV_R_aeg_RSA-2240b_2013 (1) | UP 2240 (U) | MH259261 | AchPV-2 | 74.7% | AchPV-1, AchPV-2, ThkPV-1, ThkPV-2, Sosuga | 79% |
| BatPV_R_aeg_RSA-2659_2013 (10) | UP 2659 (S), UP 2736 (S), UP 2763 (S), UP 3777 (S), UP 4169 (S), UP 6464 (S), UP 6469 (S), UPE 525 (U), UPE 809 (U), UPE 815 (U) | MH259220; MH259222; MH259224; MH259230; MH259232; MH259243; MH259244; MH259299; MH937574; MH937576 | MuV | 76.8% | MuV | 96.9% |
| BatPV_R_aeg_RSA-2752_2013 (1) | UP 2752 (S) | MH259223 | ThkPV-1 | 70.9% | ThkPV-2 | 79% |
| BatPV_R_aeg_RSA-3760a_2014 (1) | UP 3760 (S) | MH259228 | ThkPV-1, AchPV-1 | 70.4% | AchPV-2, Sosuga | 75.8% |
| BatPV_R_aeg_RSA-3777b_2014 (3) | UP 3777 (S), UP 4252 (S), UPE 087 (U) | MH259231; MH259234; MH259268 | ThkPV-1 | 73.6% | AchPV-2 | 83.8% |
| BatPV_R_aeg_RSA-4341_2014 (7) | UP 4341 (S), UP 4347 (S), UPE 118 (U), UPE 119 (U), UPE 318 (U), UPE 331 (U), UPE 343 (U) | MH259236; MH259237; MH259279; MH259280; MH259286; MH259291; MH259295 | AchPV-1 | 70.9% | ThkPV-2 | 80.6% |
| BatPV_R_aeg_RSA-MAUP4_2015 (1) | MaUP4 (U) | MH259213 | ThkPV-2 | 70.9% | AchPV-2, ThkPV-2, Sosuga | 75.8% |
| BatPV_R_aeg_RSA-UPE080_2017 (1) | UPE 080 (U) | MH259266 | ThkPV-2 | 76.3% | AchPV-1 | 83.8% |
| BatPV_R_aeg_RSA-UPE087a_2017 (2) | UPE 087 (U), UPE 195 (U) | MH259267; MH259282 | ThkPV-2 | 78.4% | Sosuga | 80.6% |
| BatPV_R_aeg_RSA-UPE092_2017 (1) | UPE 092 (U) | MH259271 | HPIV-4a | 72.5% | Sosuga | 82.2% |
| BatPV_R_aeg_RSA-UPE112b_2017 (1) | UPE 112 (U) | MH259273 | MenPV, ThkPV-1 | 72.5% | AchPV-2, Sosuga | 80.6% |
| BatPV_R_aeg_RSA-UPE122b_2017 (1) | UPE 122 (U) | MH259281 | ThkPV-1 | 73.6% | AchPV-2 | 85.4% |
| BatPV_R_aeg_RSA-UPE195b_2017 (1) | UPE 195 (U) | MH259283 | AchPV-2 | 75.2% | Sosuga | 82.2% |
| BatPV_R_aeg_RSA-UPE316_2017 (1) | UPE 316 (U) | MH259285 | AchPV-2 | 75.8% | Sosuga | 80.6% |
| BatPV_R_aeg_RSA-UPE319_2017 (2) | UPE 319 (U), UPE 481 (U) | MH259287; MH259297 | AchPV-1 | 76.3% | AchPV-1s | 85.4% |
| BatPV_R_aeg_RSA-UPE325_2017 (2) | UPE 325 (U), UPE 327 (U) | MH259288; MH259290 | ThkPV-2 | 76.3% | ThkPV-1, ThkPV-2 | 85.4% |
| BatPV_R_aeg_RSA-UPE341_2017 (1) | UPE 341 (U) | MH259294 | AchPV-2 | 76.8% | AchPV-1, AchPV-2, ThkPV-1, ThkPV-2, Sosuga | 80.6% |
| Sample | Collection Date | Li | Ki | Lu | Int | GenBank Accession Numbers |
|---|---|---|---|---|---|---|
| UP 2736 | July 2013 | − | + | − | − | MH259246 |
| UP 2763 | July 2013 | − | − | + | − | MH259247 |
| UP 3093 | September 2013 | − | − | − | + | MH259248 |
| UP 3584 | November 2013 | − | + | − | − | MH259249 |
| UP 3777 * | June 2014 | + | + | + | − | MH259275; MH259250; MH259276; MH259277 |
| UP 4169 | May 2014 | − | − | − | + | MH259214 |
| UP 4251 | June 2014 | + | + | + | + | MH259251; MH259254; MH259253; MH259252 |
| UP 4260 | June 2014 | − | + | − | − | MH259255 |
| UP 4341 | July 2014 | − | − | − | + | MH259256; |
| UP 4347 | July 2014 | − | − | − | + | MH259257 |
| UP 6464 | April 2014 | − | − | − | + | MH259258 |
| UP 6892 | June 2016 | − | + | − | − | MH259259 |
| Reference | Tested (Positive) | Positivity | Sample Type | Countries #,^ |
|---|---|---|---|---|
| Sosuga virus prevalence studies (qRT-PCR targeting the nucleoprotein of Sosuga virus) | ||||
| Amman et al., 2015 [18] | 122 (3) | 2.46% | Liver/Spleen | Uganda (various caves within the country with varying positivity) |
| 401 (3) | 0.75% | |||
| 408 (15) | 3.68% | |||
| 400 (41) | 10.25% | |||
| Rubula- and related virus detection studies (Avula–Rubulavirus specific RT-PCR assay targeting the polymerase gene) | ||||
| Drexler et al., 2012 [7] | 213 (15) | 7.04% | Spleen | Ghana, Gabon, DRC, Congo |
| Current study | 304 (29) | 9.54% | Spleen | South Africa |
| 58 (4) | 6.89% | Urine * | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortlock, M.; Dietrich, M.; Weyer, J.; Paweska, J.T.; Markotter, W. Co-Circulation and Excretion Dynamics of Diverse Rubula- and Related Viruses in Egyptian Rousette Bats from South Africa. Viruses 2019, 11, 37. https://doi.org/10.3390/v11010037
Mortlock M, Dietrich M, Weyer J, Paweska JT, Markotter W. Co-Circulation and Excretion Dynamics of Diverse Rubula- and Related Viruses in Egyptian Rousette Bats from South Africa. Viruses. 2019; 11(1):37. https://doi.org/10.3390/v11010037
Chicago/Turabian StyleMortlock, Marinda, Muriel Dietrich, Jacqueline Weyer, Janusz T. Paweska, and Wanda Markotter. 2019. "Co-Circulation and Excretion Dynamics of Diverse Rubula- and Related Viruses in Egyptian Rousette Bats from South Africa" Viruses 11, no. 1: 37. https://doi.org/10.3390/v11010037
APA StyleMortlock, M., Dietrich, M., Weyer, J., Paweska, J. T., & Markotter, W. (2019). Co-Circulation and Excretion Dynamics of Diverse Rubula- and Related Viruses in Egyptian Rousette Bats from South Africa. Viruses, 11(1), 37. https://doi.org/10.3390/v11010037