A New Cell Model for Investigating Prion Strain Selection and Adaptation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Tissue Culture
2.2. Primary Neuronal Cultures
2.3. Abbreviation of Clonal Cell Lines
2.3.1. Quantification of Prion Infection and Rates of Prion Replication
2.3.2. Quantification of Prion Titers Using the Scrapie Cell Assay in Endpoint Format (SCEPA)
2.4. Single Cell Cloning and Cryopreservation
2.5. Preparation of Cell Homogenates
2.6. Proteinase K Digestion of Cell and Brain Homogenates
2.6.1. Cell Homogenates
2.6.2. Brain Homogenates
2.7. Western Blotting
2.8. Laser-Scanning Microscopy
2.9. Determination of Rod Length in Primary Neuronal Cultures
3. Results
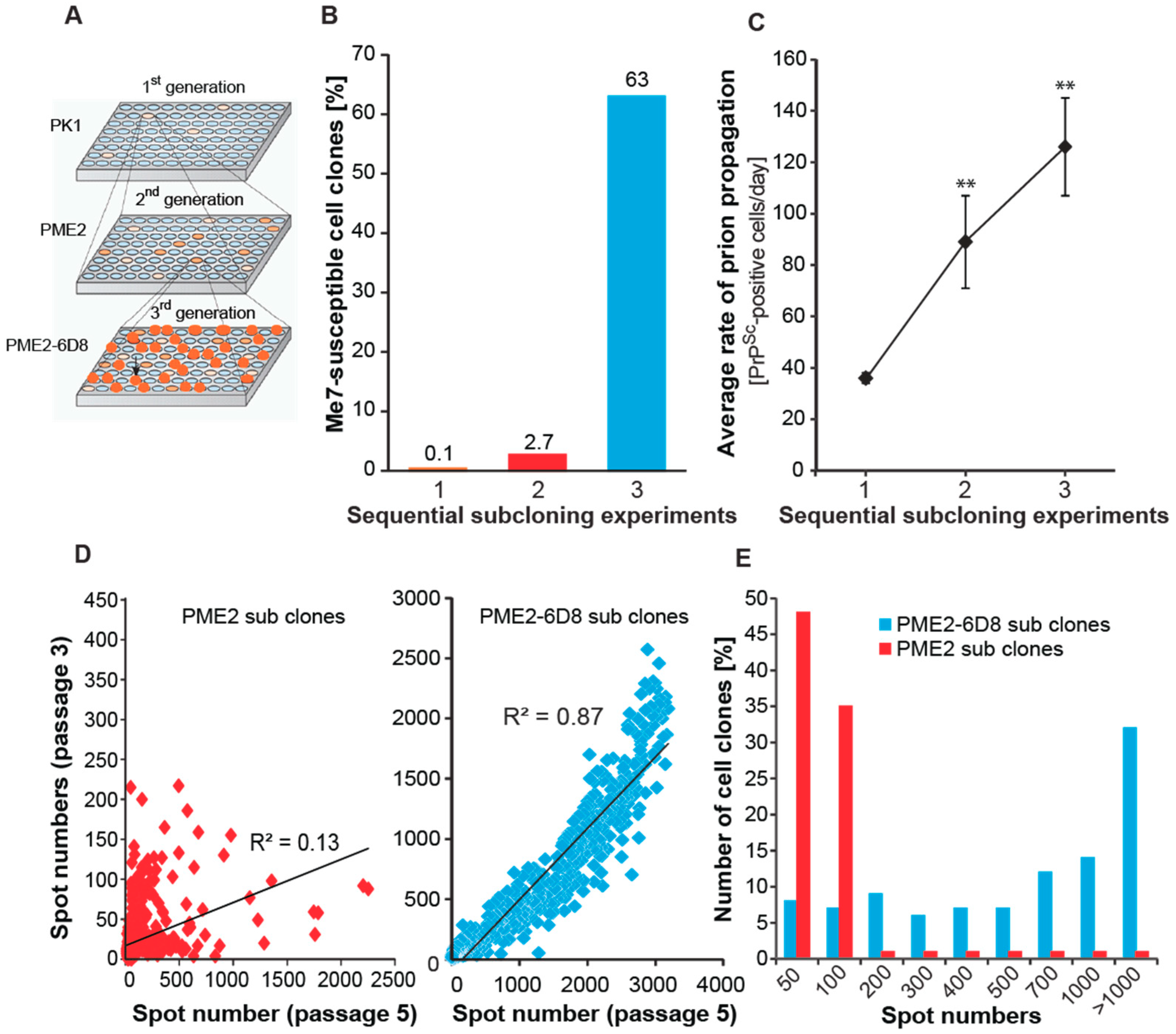
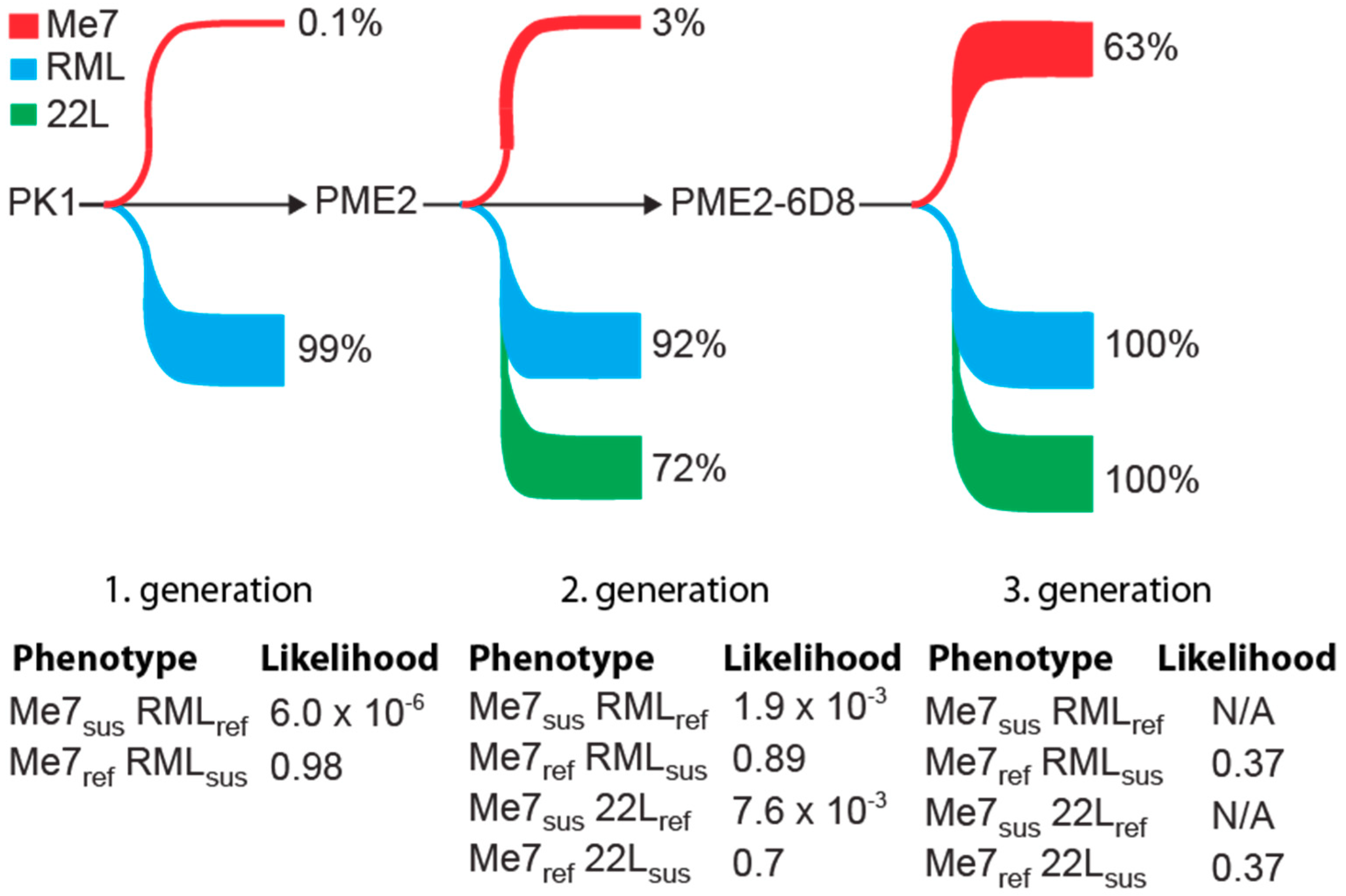
3.1. Isolation of Two Rare PK1 Cell Clones Potentially Permissive to Me7
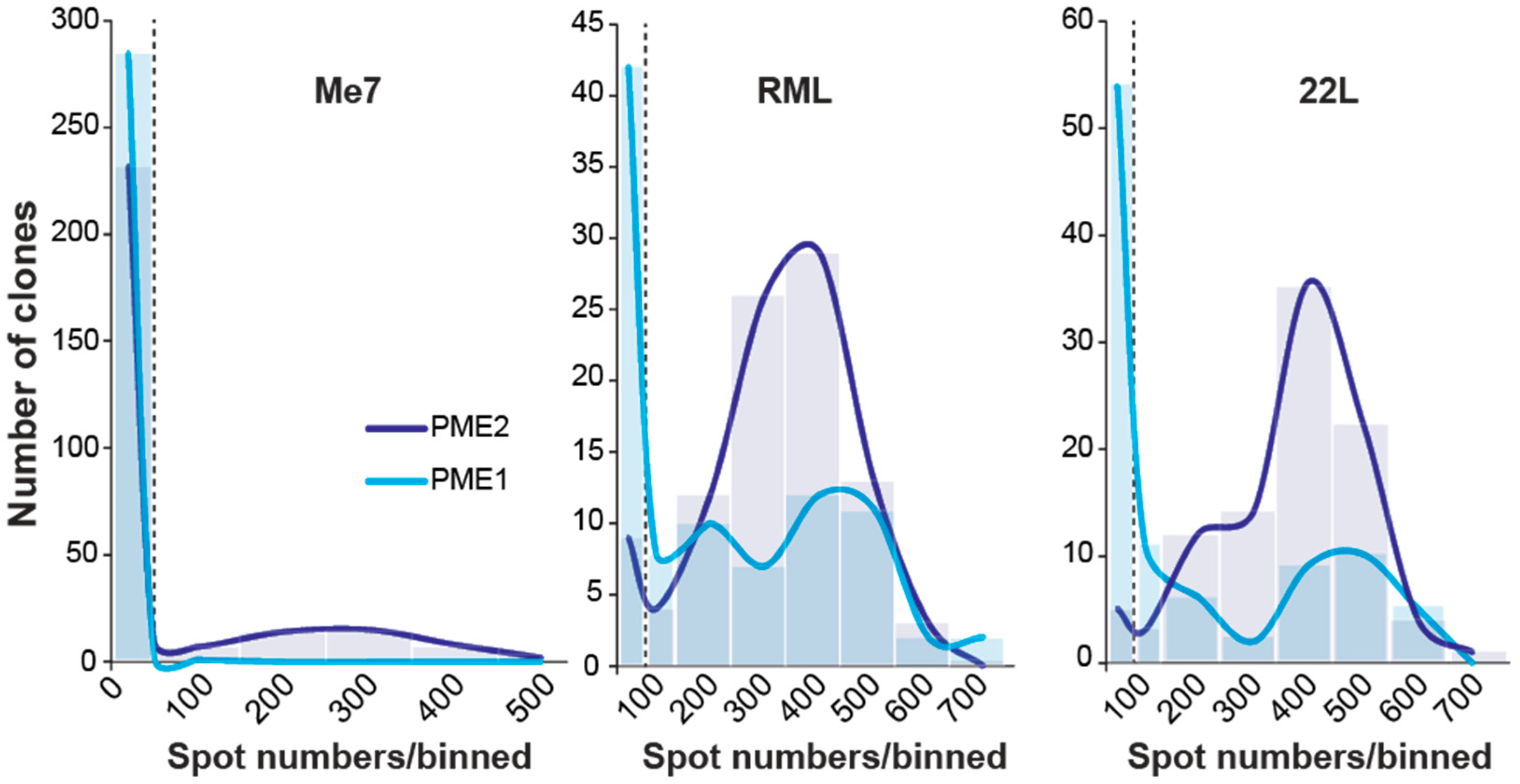
3.2. Progressive Enrichment of Me7-Susceptible Cells by Subcloning
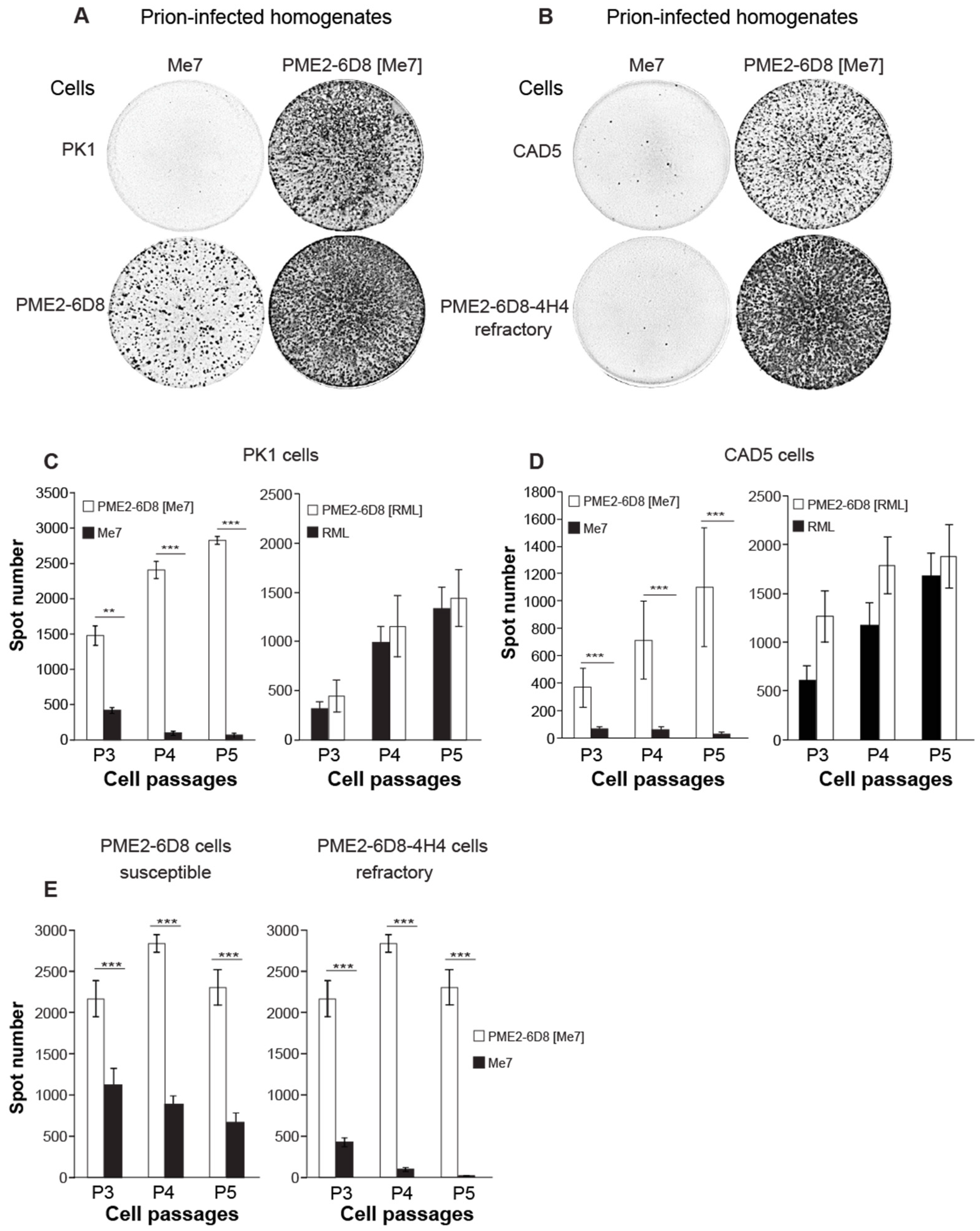
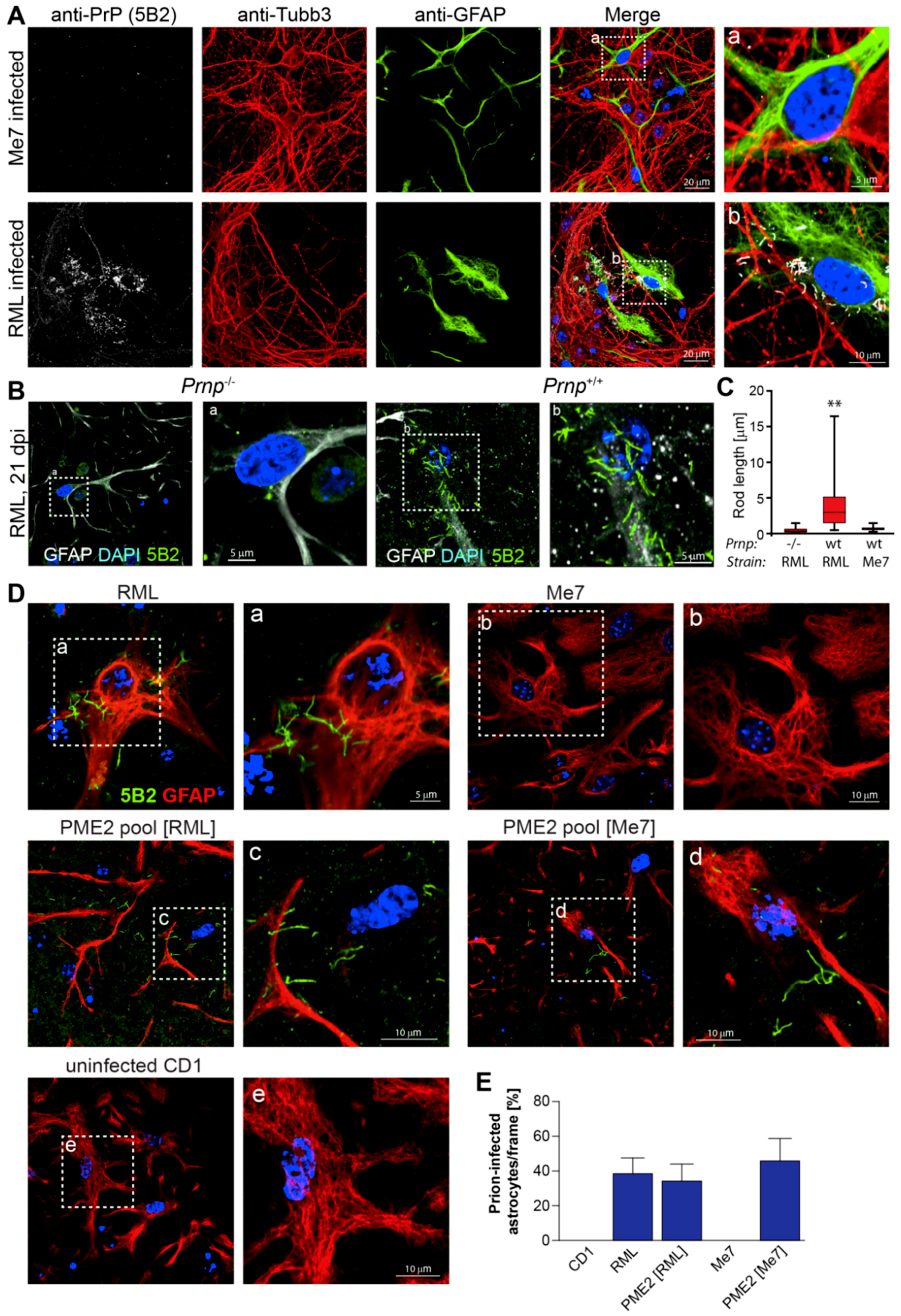
3.3. Me7-Infected PME2 Cells Deposit PrPd Aggregates and Differ in Their PK Sensitivity from RML-Infected Cells
3.4. Evidence of Prion Strain Adaptation in Me7-Susceptible PME2 Clones
3.5. Expanded Host Range of Cell-Adapted Me7 Prions in Primary Neuronal Cells
4. Discussion
4.1. A Bottom-Up Approach to Study Selective Neuronal Vulnerability Using Strain-Selective Isogenic Neuronal Cells
4.2. Prion Strain Variation and Adaptation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Griffith, J.S. Self Replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Molecular Biology of Prion Diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, H.A. Neuropathology of human prion diseases (spongiform encephalopathies). Dev. Biol. Stand 1993, 80, 71–90. [Google Scholar] [PubMed]
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s disease: Genetics and pathogenesis. Annu. Rev. Pathol. 2011, 6, 193–222. [Google Scholar] [CrossRef]
- Gallassi, R.; Morreale, A.; Montagna, P.; Gambetti, P.; Lugaresi, E. “Fatal familial insomnia”: Neuropsychological study of a disease with thalamic degeneration. Cortex 1992, 28, 175–187. [Google Scholar] [CrossRef]
- Almer, G.; Hainfellner, J.A.; Brücke, T.; Jellinger, K.; Kleinert, R.; Bayer, G.; Windl, O.; Kretzschmar, H.A.; Hill, A.; Sidle, K.; et al. Fatal familial insomnia: A new Austrian family. Brain 1999, 122, 5–16. [Google Scholar] [CrossRef]
- Guentchev, M.; Wanschitz, J.; Voigtlander, T.; Flicker, H.; Budka, H. Selective neuronal vulnerability in human prion diseases. Fatal familial insomnia differs from other types of prion diseases. Am. J. Pathol. 1999, 155, 1453–1457. [Google Scholar] [CrossRef]
- Rubenstein, R.; Deng, H.; Race, R.E.; Ju, W.; Scalici, C.L.; Papini, M.C.; Kascsak, R.J.; Carp, R.I. Demonstration of scrapie strain diversity in infected PC12 cells. J. Gen. Virol. 1992, 73, 3027–3031. [Google Scholar] [CrossRef]
- Vorberg, I.; Raines, A.; Priola, S.A. Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro. J. Biol. Chem. 2004. [Google Scholar] [CrossRef] [PubMed]
- Mahal, S.P.; Baker, C.A.; Demczyk, C.A.; Smith, E.W.; Julius, C.; Weissmann, C. Prion strain discrimination in cell culture: The cell panel assay. Proc. Natl. Acad Sci. USA 2007, 104, 20908–20913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef] [PubMed]
- Fehlinger, A.; Wolf, H.; Hossinger, A.; Duernberger, Y.; Pleschka, C.; Riemschoss, K.; Liu, S.; Bester, R.; Paulsen, L.; Priola, S.A.; et al. Prion strains depend on different endocytic routes for productive infection. Sci. Rep. 2017, 7, 6923. [Google Scholar] [CrossRef] [PubMed]
- Marbiah, M.M.; Harvey, A.; West, B.T.; Louzolo, A.; Banerjee, P.; Alden, J.; Grigoriadis, A.; Hummerich, H.; Kan, H.M.; Cai, Y.; et al. Identification of a gene regulatory network associated with prion replication. EMBO J. 2014, e201387150. [Google Scholar] [CrossRef] [PubMed]
- Bosque, P.J.; Prusiner, S.B. Cultured cell sublines highly susceptible to prion infection. J. Virol. 2000, 74, 4377–4386. [Google Scholar] [CrossRef] [PubMed]
- Klohn, P.; Stoltze, L.; Flechsig, E.; Enari, M.; Weissmann, C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad Sci. USA 2003, 100, 11666–11671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian evolution of prions in cell culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef]
- Thackray, A.M.; Klein, M.A.; Aguzzi, A.; Bujdoso, R. Chronic subclinical prion disease induced by low-dose inoculum. J. Virol. 2002, 76, 2510–2517. [Google Scholar] [CrossRef]
- Thackray, A.M.; Hopkins, L.; Klein, M.A.; Bujdoso, R. Mouse-adapted ovine scrapie prion strains are characterized by different conformers of PrPSc. J. Virol. 2007, 81, 12119–12127. [Google Scholar] [CrossRef]
- Weissmann, C. The state of the prion. Nat. Rev. Microbiol. 2004, 2, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Zanusso, G.; Liu, D.; Ferrari, S.; Hegyi, I.; Yin, X.; Aguzzi, A.; Hornemann, S.; Liemann, S.; Glockshuber, R.; Manson, J.C.; et al. Prion protein expression in different species: Analysis with a panel of new mAbs. Proc. Natl. Acad Sci. USA 1998, 95, 8812–8816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arima, K.; Nishida, N.; Sakaguchi, S.; Shigematsu, K.; Atarashi, R.; Yamaguchi, N.; Yoshikawa, D.; Yoon, J.; Watanabe, K.; Kobayashi, N.; et al. Biological and biochemical characteristics of prion strains conserved in persistently infected cell cultures. J. Virol. 2005, 79, 7104–7112. [Google Scholar] [CrossRef] [PubMed]
- Arjona, A.; Simarro, L.; Islinger, F.; Nishida, N.; Manuelidis, L. Two Creutzfeldt-Jakob disease agents reproduce prion protein-independent identities in cell cultures. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef] [PubMed]
- Dichter, M.A. Rat cortical neurons in cell culture: Culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res. 1978, 149, 279–293. [Google Scholar] [CrossRef]
- Li, R.L.; Liu, T.; Wong, B.S.; Pan, T.; Morillas, M.; Swietnicki, W.; O’Rourke, K.; Gambetti, P.; Surewicz, W.K.; Sy, M.S. Identification of an epitope in the C terminus of normal prion protein whose expression is modulated by binding events in the N terminus. J. Mol. Biol. 2000, 301, 567–573. [Google Scholar] [CrossRef]
- Rouvinski, A.; Karniely, S.; Kounin, M.; Moussa, S.; Goldberg, M.D.; Warburg, G.; Lyakhovetsky, R.; Papy-Garcia, D.; Kutzsche, J.; Korth, C.; et al. Live imaging of prions reveals nascent PrPSc in cell-surface, raft-associated amyloid strings and webs. J. Cell Biol. 2014, 204, 423–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Li, R.; Kang, S.C.; Wong, B.S.; Wisniewski, T.; Sy, M.S. Epitope scanning reveals gain and loss of strain specific antibody binding epitopes associated with the conversion of normal cellular prion to scrapie prion. J. Neurochem. 2004, 90, 1205–1217. [Google Scholar] [CrossRef]
- Clarke, A.R.; Jackson, G.S.; Collinge, J. The molecular biology of prion propagation. Philos. Trans. R. Soc. B Boil. Sci. 2001, 356, 185–194. [Google Scholar]
- Gambetti, P.; Parchi, P.; Chen, S.G. Hereditary Creutzfeldt-Jakob disease and fatal familial insomnia. Clin. Lab. Med. 2003, 23, 43–64. [Google Scholar] [CrossRef]
- Wadsworth, J.D.; Collinge, J. Molecular pathology of human prion disease. Acta Neuropathol. 2011, 121, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Dickinson, A.G. The sequential development of the brain lesion of scrapie in three strains of mice. J. Comp. Pathol. 1968, 78, 301–311. [Google Scholar] [CrossRef]
- Wells, G.A.H.; Wilesmith, J.W. The neuropathology and epidemiology of bovine spongiform encephalopathy. Brain Pathol. 1995, 5, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Groschup, M.H.; Kuczius, T.; Junghans, F.; Sweeney, T.; Bodemer, W.; Buschmann, A. Characterization of BSE and scrapie strains/isolates. In Prion Diseases; Springer: Vienna, Austria, 2000; pp. 217–226. [Google Scholar]
- Zerr, I.; Poser, S. Clinical diagnosis and differential diagnosis of CJD and vCJD. With special emphasis on laboratory tests. APMIS 2002, 110, 88–98. [Google Scholar] [CrossRef]
- Gonzalez, L.; Martin, S.; Jeffrey, M. Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie- and BSE-infected sheep: Implications for differential cell targeting and PrP processing. J. Gen. Virol. 2003, 84, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Vorberg, I.; Raines, A.; Story, B.; Priola, S.A. Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J. Infect. Dis. 2004, 189, 431–439. [Google Scholar] [CrossRef]
- Solassol, J.; Crozet, C.; Lehmann, S. Prion propagation in cultured cells. Br. Med. Bull. 2003, 66, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Vilette, D. Cell models of prion infection. Vet. Res. 2008, 39, 10. [Google Scholar] [CrossRef]
- Kosti, I.; Jain, N.; Aran, D.; Butte, A.J.; Sirota, M. Cross-tissue Analysis of Gene and Protein Expression in Normal and Cancer Tissues. Sci. Rep. 2016, 6, 24799. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.A.; Anderson, C.R.; Terkildsen, S.E.; Davis, R.C.; Pack, L.D.; Bhargava, S.; Clarke, H.R.G. Antibody expression stability in CHO clonally derived cell lines and their subclones: Role of methylation in phenotypic and epigenetic heterogeneity. Biotechnol. Prog. 2018, 34, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Pilbrough, W.; Munro, T.P.; Gray, P. Intraclonal protein expression heterogeneity in recombinant CHO cells. PLoS ONE 2009, 4, e8432. [Google Scholar] [CrossRef] [PubMed]
- Buganim, Y.; Faddah, D.A.; Cheng, A.W.; Itskovich, E.; Markoulaki, S.; Ganz, K.; Klemm, S.L.; van Oudenaarden, A.; Jaenisch, R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 2012, 150, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Papp, B.; Plath, K. Epigenetics of reprogramming to induced pluripotency. Cell 2013, 152, 1324–1343. [Google Scholar] [CrossRef] [PubMed]
- Munsky, B.; Neuert, G.; van Oudenaarden, A. Using gene expression noise to understand gene regulation. Science 2012, 336, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Sidle, K.C.; Meads, J.; Ironside, J.; Hill, A.F. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996, 383, 685–690. [Google Scholar] [CrossRef]
- Parchi, P.; Castellani, R.; Capellari, S.; Ghetti, B.; Young, K.; Chen, S.G.; Farlow, M.; Dickson, D.W.; Sims, A.A.F.; Trojanowski, J.Q.; et al. Molecular Basis of Phenotypic Variability in Sporadic Creutzfeldt-Jakob Disease. Ann. Neurol. 1996, 39, 669–680. [Google Scholar] [CrossRef]
- Everest, S.J.; Thorne, L.; Barnicle, D.A.; Edwards, J.C.; Elliott, H.; Jackman, R.; Hope, J. Atypical prion protein in sheep brain collected during the British scrapie-surveillance programme. J. Gen. Virol. 2006, 87, 471–477. [Google Scholar] [CrossRef]
- Pirisinu, L.; Nonno, R.; Esposito, E.; Benestad, S.L.; Gambetti, P.; Agrimi, U.; Zou, W.Q. Small ruminant nor98 prions share biochemical features with human gerstmann-straussler-scheinker disease and variably protease-sensitive prionopathy. PLoS ONE 2013, 8, e66405. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Stoeck, K.; Glatzel, M.; Vey, M.; Bellon, A.; Aguzzi, A. Coexistence of multiple PrP(Sc) types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol. 2005, 4, 805–814. [Google Scholar] [CrossRef]
- Lloyd, S.E.; Linehan, J.M.; Desbruslais, M.; Joiner, S.; Buckell, J.; Brandner, S.; Wadsworth, J.D.F.; Collinge, J. Characterization of two distinct prion strains derived from bovine spongiform encephalopathy transmissions to inbred mice. J. Gen. Virol. 2004, 85, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Beringue, V.; Andreoletti, O.; Le Dur, A.; Essalmani, R.; Vilotte, J.L.; Lacroux, C.; Reine, F.; Herzog, L.; Biacabe, A.G.; Baron, T.; et al. A bovine prion acquires an epidemic bovine spongiform encephalopathy strain-like phenotype on interspecies transmission. J. Neurosci. 2007, 27, 6965–6971. [Google Scholar] [CrossRef] [PubMed]
- Cancellotti, E.; Barron, R.M.; Bishop, M.T.; Hart, P.; Wiseman, F.; Manson, J.C. The role of host PrP in Transmissible Spongiform Encephalopathies. Biochim. Biophys. Acta 2007, 1772, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groschup, M.H.; Buschmann, A. Rodent models for prion diseases. Vet. Res. 2008, 39, 32. [Google Scholar] [CrossRef]
- Parchi, P.; Strammiello, R.; Giese, A.; Kretzschmar, H. Phenotypic variability of sporadic human prion disease and its molecular basis: Past, present, and future. Acta Neuropathol. 2010. [Google Scholar] [CrossRef]
- Baskakov, I.V.; Katorcha, E. Multifaceted Role of Sialylation in Prion Diseases. Front. Neurosci. 2016, 10, 358. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PK1 Subclones | Spot Number |
|---|---|
| PME2 | 378 |
| PME1 | 351 |
| 16 clones | 100–200 |
| 42 clones | 50–100 |
| 660 clones | 0–50 |
| Scrapie Cell Assay | ||
|---|---|---|
| PME2 Subclones (Round 1) | PrPSc-Infected Cells (Passage 3) | PrPSc-Infected Cells (Passage 5) |
| 1H10 | 88 | 2235 |
| 1F5 | 92 | 2206 |
| 1B2 | 59 | 1748 |
| 3B3 | 58 | 1788 |
| 6D8 | 98 | 1356 |
| 2G6 | 49 | 1229 |
| 2E9 | 20 | 1287 |
| 1A8 | 7 | 10 |
| 5F12 | 13 | 11 |
| PME2-6D8 Subclones (Round 2) | PrPSc-Infected Cells (Passage 3) | PrPSc-Infected Cells (Passage 5) |
| 4F11 | 2079 | 3191 |
| 2C6 | 1863 | 3169 |
| 5B9 | 2178 | 3152 |
| 5B1 | 1618 | 3147 |
| 5F1 | 2131 | 3142 |
| 1G1 | 1844 | 3121 |
| 5E1 | 1997 | 3103 |
| 5C2 | 2030 | 3071 |
| PME2 | 41 ± 8 | 137 ± 42 |
| Number of PrPSc-Infected Cells after Infection with | |||
|---|---|---|---|
| Me7-Refractory PME2-6D8 Subclones | Me7 | 22L | RML |
| 5E6 | 3 | 3038 | 2881 |
| 4C7 | 4 | 3151 | 2910 |
| 4D4 | 5 | 3170 | 2937 |
| 4C12 | 6 | 2786 | 2724 |
| 5G5 | 6 | 2949 | 2851 |
| 5E2 | 7 | 2935 | 2656 |
| 4H4 | 8 | 2723 | 2675 |
| Me7-Susceptible PME2-6D8 Subclones | Me7 | 22L | RML |
| 4F11 | 3191 | 2849 | 2883 |
| 2C6 | 3169 | 2858 | 2763 |
| 5B9 | 3152 | 2898 | 2824 |
| 5B1 | 3147 | 2772 | 3064 |
| 5F1 | 3142 | 2420 | 2368 |
| 1G1 | 3121 | 2602 | 2293 |
| 5E1 | 3103 | 2576 | 2840 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Philiastides, A.; Ribes, J.M.; Yip, D.C.-M.; Schmidt, C.; Benilova, I.; Klöhn, P.-C. A New Cell Model for Investigating Prion Strain Selection and Adaptation. Viruses 2019, 11, 888. https://doi.org/10.3390/v11100888
Philiastides A, Ribes JM, Yip DC-M, Schmidt C, Benilova I, Klöhn P-C. A New Cell Model for Investigating Prion Strain Selection and Adaptation. Viruses. 2019; 11(10):888. https://doi.org/10.3390/v11100888
Chicago/Turabian StylePhiliastides, Alexandra, Juan Manuel Ribes, Daniel Chun-Mun Yip, Christian Schmidt, Iryna Benilova, and Peter-Christian Klöhn. 2019. "A New Cell Model for Investigating Prion Strain Selection and Adaptation" Viruses 11, no. 10: 888. https://doi.org/10.3390/v11100888
APA StylePhiliastides, A., Ribes, J. M., Yip, D. C.-M., Schmidt, C., Benilova, I., & Klöhn, P.-C. (2019). A New Cell Model for Investigating Prion Strain Selection and Adaptation. Viruses, 11(10), 888. https://doi.org/10.3390/v11100888