Immunogenicity and Efficacy of a Novel Multi-Antigenic Peptide Vaccine Based on Cross-Reactivity between Feline and Human Immunodeficiency Viruses

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Immune Analyses Used in Selecting Protective T-Cell Epitopes on FIV
2.2. The Approaches for Selecting Mutation-Resistant, Protective FIV Epitopes
2.3. In Sillico HLA Algorithms
2.4. Constructing the MAP Vaccine Immunogens into A Vaccine
2.5. Animals
2.6. MAP Vaccines and Vaccination
2.7. Monitoring Vaccine Immunogenicity
2.8. FIV Challenge and Monitoring FIV Infection
2.9. Statistical Analyses and Protection Rate
3. Results
3.1. Selection of Mutation-Resistant, Protective T-cell Epitopes on FIV
3.2. Designing the Vaccine Immunogens
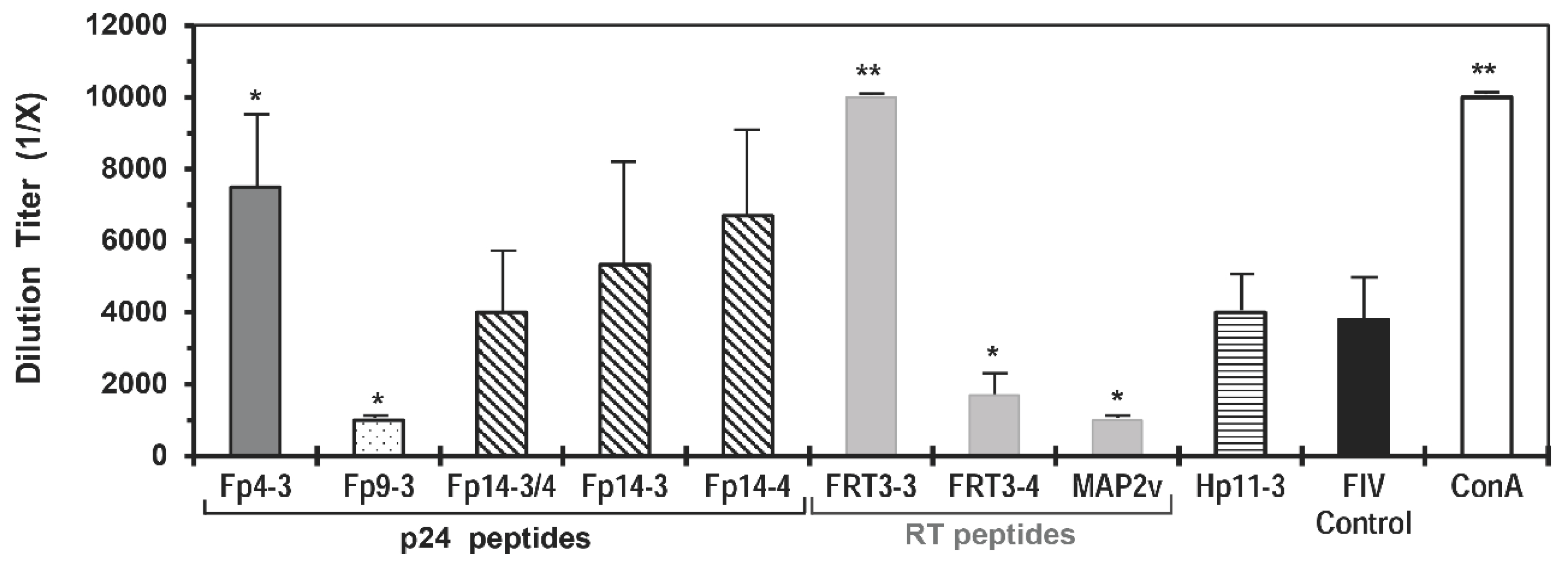
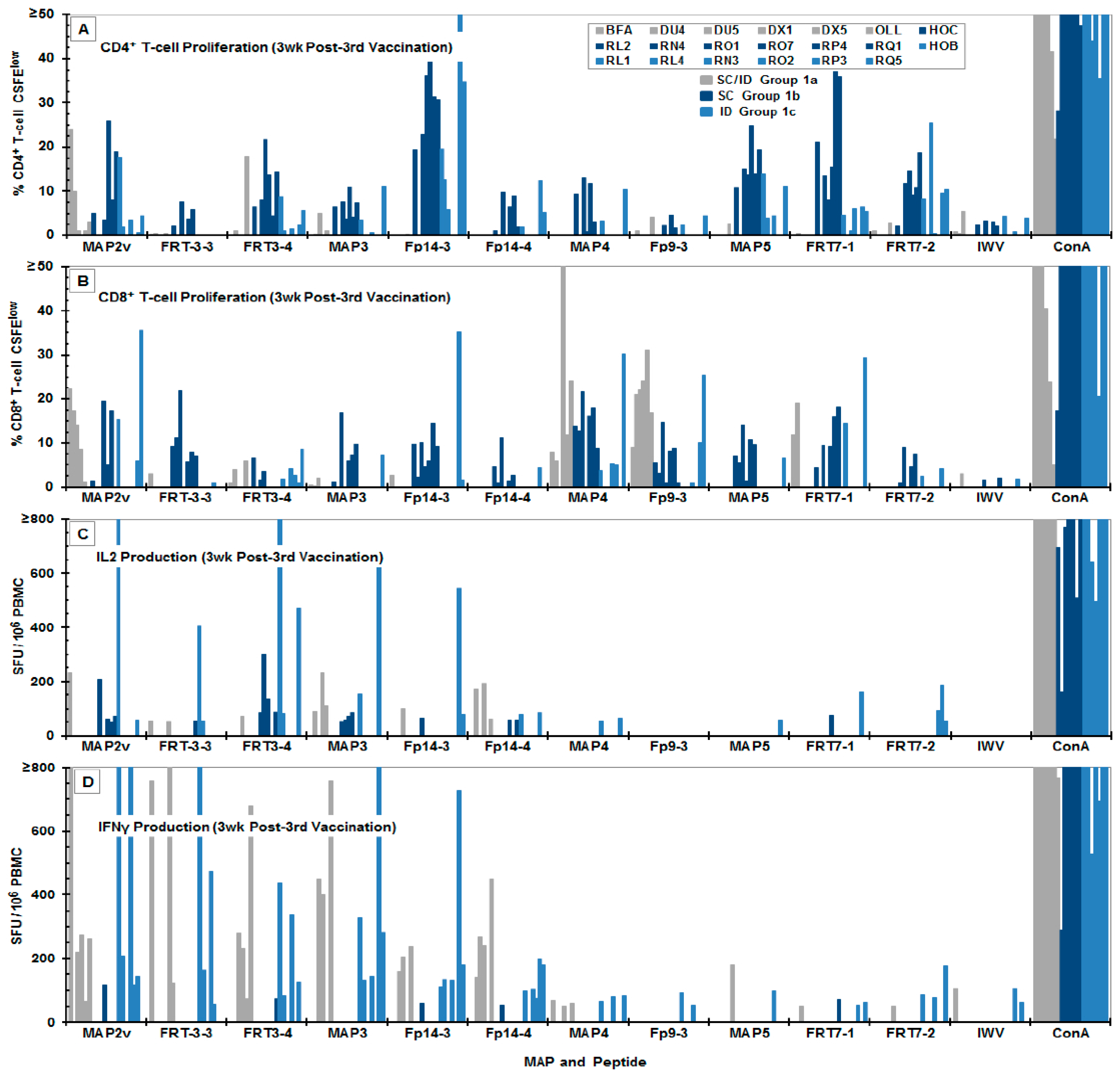
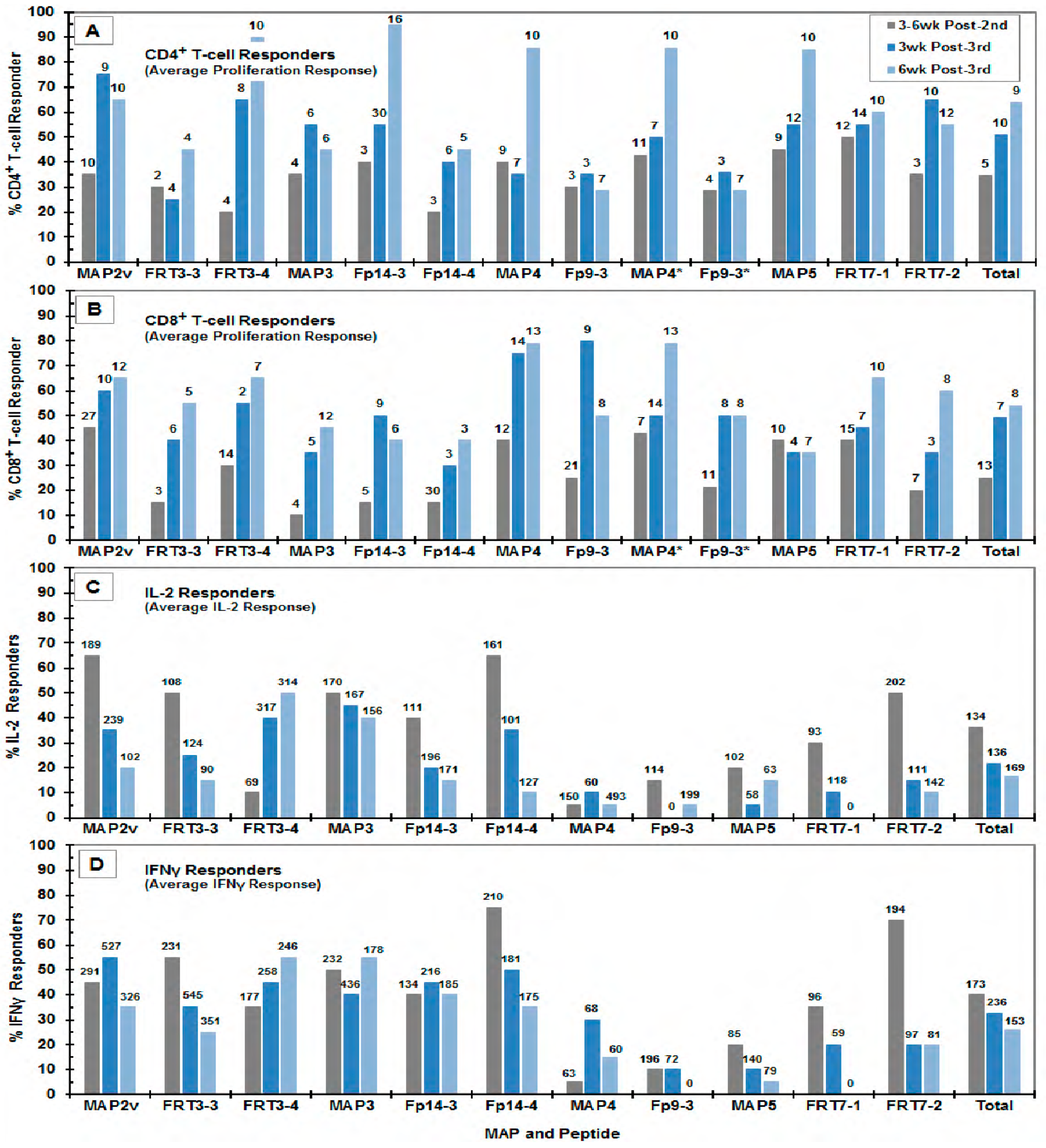
3.3. Immunogenicity of the MAP Vaccines
3.4. MAP Vaccine Efficacy against Challenge with Pathogenic Subtype B FIV (FIVFC1)
3.5. Statistical Comparison between the Unprotected/Vaccinated and the Protected/Vaccinated Cats
4. Discussion
4.1. The Selection of Potential Protective HIV Peptides
4.2. The in vitro Selection of Protective FIV Peptides Compared to the Peptide-Specific Immunity of Vaccinated Cats
4.3. MAP Vaccine Immunity and Efficacy
4.4. Correlates of MAP Vaccine Protection
4.5. Future Directions in Improving T Cell-Based FIV/HIV Vaccines
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cohen, K.W.; Frahm, N. Current views on the potential for development of a HIV vaccine. Expert Opin. Biol. Ther. 2017, 17, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Seddiki, N.; Lévy, Y. Therapeutic HIV-1 vaccine: Time for immunomodulation and combinatorial strategies. Curr. Opin. HIV AIDS 2018, 13, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Wijesundara, D.K.; Ranasinghe, C.; Grubor-Bauk, B.; Gowans, E.J. Emerging targets for developing T cell-mediated vaccines for human immunodeficiency virus (HIV)-1. Front. Microbiol. 2017, 8, 2091. [Google Scholar] [CrossRef] [PubMed]
- Sahay, B.; Nguyen, C.Q.; Yamamoto, J.K. Conserved HIV epitopes for an effective HIV vaccine. J. Clin. Cell. Immunol. 2017, 8, pii:518. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. MOPH-TAVEG Investigators. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Gilbert, P.B.; McElrath, M.J.; Zolla-Pazner, S.; Tomaras, G.D.; Alam, S.M.; Evans, D.T.; Montefiori, D.C.; Karnasuta, C.; Sutthent, R.; et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 2012, 366, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- de Souza, M.S.; Ratto-Kim, S.; Chuenarom, W.; Schuetz, A.; Chantakulkij, S.; Nuntapinit, B.; Valencia-Micolta, A.; Thelian, D.; Nitayaphan, S.; Pitisuttithum, P.; et al. Ministry of Public Health-Thai AIDS Vaccine Evaluation Group Collaborators. The Thai phase III trial (RV144) vaccine regimen induces T cell responses that preferentially target epitopes within the V2 region of HIV-1 envelope. J. Immunol. 2012, 188, 5166–5176. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Piatak, M., Jr.; Ventura, A.B.; Hughes, C.M.; Gilbride, R.M.; Ford, J.C.; Oswald, K.; Shoemaker, R.; Li, Y.; Lewis, M.S.; et al. Immune clearance of highly pathogenic SIV infection. Nature 2013, 502, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.K.; Pu, R.; Martin, M.M.; Noon-Song, E.N.; Zwijnenberg, R.; Yamamoto, J.K. Feline immunodeficiency virus (FIV) vaccine efficacy and FIV neutralizing antibodies. Vaccine 2014, 32, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Westman, M.E.; Malik, R.; Hall, E.; Harris, M.; Norris, J.M. The protective rate of the feline immunodeficiency virus vaccine: An Australian field study. Vaccine 2016, 34, 4752–4758. [Google Scholar] [CrossRef]
- Hansen, S.G.; Sacha, J.B.; Hughes, C.M.; Ford, J.C.; Burwitz, B.J.; Scholz, I.; Gilbride, R.M.; Lewis, M.S.; Gilliam, A.N.; Ventura, A.B.; et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 2013, 340, 1237874-1–1237874-17. [Google Scholar] [CrossRef] [PubMed]
- Aranyos, A.M.; Roff, S.R.; Pu, R.; Owen, J.L.; Coleman, J.K.; Yamamoto, J.K. An initial examination of the potential role of T-cell immunity in protection against feline immunodeficiency virus (FIV) infection. Vaccine 2016, 34, 1480–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahay, B.; Yamamoto, J.K. Lessons learned in developing a commercial FIV vaccine: The immunity required for an effective HIV-1 vaccine. Viruses 2018, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Omori, M.; Pu, R.; Tanabe, T.; Hou, W.; Coleman, J.K.; Arai, M.; Yamamoto, J.K. Cellular immune responses to feline immunodeficiency virus (FIV) induced by dual-subtype FIV vaccine. Vaccine 2004, 23, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K. Clinical aspects of feline immunodeficiency and feline leukemia virus infection. Vet. Immunol. Immunopathol. 2011, 143, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Sahay, B.; Aranyos, A.M.; McAvoy, A.; Yamamoto, J.K. Utilization of feline ELISpot to evaluate the immunogenicity of a T cell-based FIV MAP vaccine. Methods Mol. Biol. 2018, 1808, 197–219. [Google Scholar] [CrossRef] [PubMed]
- Sanou, M.P.; Roff, S.R.; Mennella, A.; Sleasman, J.W.; Rathore, M.H.; Yamamoto, J.K.; Levy, J.A. Evolutionarily conserved epitopes on human immunodeficiency virus type 1 (HIV-1) and feline immunodeficiency virus reverse transcriptases detected by HIV-1-infected subjects. J. Virol. 2013, 87, 10004–10015. [Google Scholar] [CrossRef] [PubMed]
- Roff, S.R.; Sanou, M.P.; Rathore, M.H.; Levy, J.A.; Yamamoto, J.K. Conserved epitopes on HIV-1, FIV and SIV p24 proteins are recognized by HIV-1 infected subjects. Hum. Vaccin. Immunother. 2015, 11, 1540–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, J.R.; Sanou, M.P.; Coleman, J.K.; Yamamoto, J.K. Evolutionarily conserved T-cell epitopes on FIV for designing an HIV/AIDS vaccine. Vet. Immunol. Immunopathol. 2011, 143, 246–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Net-MHC 4.0 Server, DTU Bioinformatics. Available online: http://www.cbs.dtu.dk/services/NetMHC/ (accessed on 22 August 2018).
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Net-MHCII 2.3 Server. DTU Bioinformatics. Available online: http://www.cbs.dtu.dk/services/NetMHCII/ (accessed on 23 August 2018).
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef] [PubMed]
- NetCTL 1.2 Server, DTU Bioinformatics. Available online: http://www.cbs.dtu.dk/services/NetCTL/ (accessed on 26 August 2018).
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Uhl, E.W.; Heaton-Jones, T.G.; Pu, R.; Yamamoto, J.K. FIV vaccine development and its importance to veterinary and human medicine: A review FIV vaccine 2002 update and review. Vet. Immunol. Immunopathol. 2002, 90, 113–132. [Google Scholar] [CrossRef]
- BenMohamed, L.; Wechsler, S.L.; Nesburn, A.B. Lipopeptide vaccines—Yesterday, today, and tomorrow. Lancet Infect. Dis. 2002, 2, 425–431. [Google Scholar] [CrossRef]
- Zhang, X.; Dervillez, X.; Chentoufi, A.A.; Badakhshan, T.; Bettahi, I.; Benmohamed, L. Targeting the genital tract mucosa with a lipopeptide/recombinant adenovirus prime/boost vaccine induces potent and long-lasting CD8+ T cell immunity against herpes: Importance of MyD88. J. Immunol. 2012, 189, 4496–4509. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Hruska, K.A.; Dowdy, S.F. Protein transduction: Unrestricted delivery into all cells? Trends Cell Biol. 2000, 10, 290–295. [Google Scholar] [CrossRef]
- Marshall BioResources, Liberty Research Inc., Antibody Profile Defined/Specific Pathogen Free Domestic Shorthair Cats. Available online: https://www.marshallbio.com/apdspf-cats (accessed on 6 January 2019).
- Yamamoto, J.K.; Pu, R.; Sato, E.; Hohdatsu, T. Feline immunodeficiency virus pathogenesis and development of a dual-subtype feline-immunodeficiency-virus vaccine. AIDS 2007, 21, 547–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Los Alamos National Laboratory (LANL), QuickAlign. Available online: https://www.hiv.lanl.gov/content/sequence/QUICK_ALIGNv2/QuickAlign.html (accessed on 30 October 2018).
- Okada, S.; Pu, R.; Young, E.; Stoffs, W.V.; Yamamoto, J.K. Superinfection of cats with feline immunodeficiency virus subtypes A and B. AIDS Res. Hum. Retroviruses. 1994, 10, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.K.; Barré-Sinoussi, F.; Bolton, V.; Pedersen, N.C.; Gardner, M.B. Human alpha- and beta-interferon but not gamma- suppress the in vitro replication of LAV, HTLV-III, and ARV-2. J. Interferon Res. 1986, 6, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.A.; Spire, B.; Dormont, D.; Barre-Sinoussi, F.; Montagnier, L.; Chermann, J.C. Characterization of the RNA dependent DNA polymerase of a new human T-lymphotropic retrovirus (lymphadenopathy associated virus). Biochem. Biophys. Res. Commun. 1984, 121, 126–133. [Google Scholar] [CrossRef]
- Arai, M.; Earl, D.D.; Yamamoto, J.K. Is AZT/3TC therapy effective against FIV infection or immunopathogenesis? Vet. Immunol. Immunopathol. 2002, 85, 189–204. [Google Scholar] [CrossRef]
- Douek, D.C.; Roederer, M.; Koup, R.A. Emerging concepts in the immunopathogenesis of AIDS. Annu. Rev. Med. 2009, 60, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, P.; Hawkins, N.; Dinges, W.L.; Liu, Y.; Gabriel, E.E.; Swan, D.A.; Stevens, C.E.; Maenza, J.; Collier, A.C.; Mullins, J.I.; et al. Superior control of HIV-1 replication by CD8+ T cells targeting conserved epitopes: Implications for HIV vaccine design. PLoS ONE 2013, 8, e64405. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Cirión, A.; Sinet, M.; Shin, S.Y.; Urrutia, A.; Versmisse, P.; Lacabaratz, C.; Boufassa, F.; Avettand-Fènoël, V.; Rouzioux, C.; Delfraissy, J.F.; et al. ANRS EP36 HIV Controllers Study Group. Heterogeneity in HIV suppression by CD8 T cells from HIV controllers: Association with Gag-specific CD8 T cell responses. J. Immunol. 2009, 182, 7828–7837. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.H.; Bansal, A.; Sabbaj, S.; Bakari, J.; Mulligan, M.J.; Goepfert, P.A. Magnitude of functional CD8+ T-cell responses to the gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J. Virol. 2002, 76, 2298–2305. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.R.; van Baalen, C.A.; Holwerda, A.M.; Kerkhof Garde, S.R.; Bende, R.J.; Keet, I.P.; Eeftinck-Schattenkerk, J.K.; Osterhaus, A.D.; Schuitemaker, H.; Miedema, F. Kinetics of Gag-specific cytotoxic T lymphocyte responses during the clinical course of HIV-1 infection: A longitudinal analysis of rapid progressors and long-term asymptomatics. J. Exp. Med. 1995, 181, 1365–1372. [Google Scholar] [CrossRef]
- World Health Organization, Global Health Observatory (GHO) data. Available online: https://www.who.int/gho/hiv/epidemic_status/en/ (accessed on 9 January 2019).
- Siliciano, J.D.; Siliciano, R.F. Recent developments in the effort to cure HIV infection: Going beyond N = 1. J. Clin. Investig. 2016, 126, 409–414. [Google Scholar] [CrossRef]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. Department of Health and Human Services. Available online: https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf (accessed on 10 January 2019).
- Gottardo, R.; Bailer, R.T.; Korber, B.T.; Gnanakaran, S.; Phillips, J.; Shen, X.; Tomaras, G.D.; Turk, E.; Imholte, G.; Eckler, L.; et al. Plasma IgG to linear epitopes in the V2 and V3 regions of HIV-1 gp120 correlate with a reduced risk of infection in the RV144 vaccine efficacy trial. PLoS ONE. 2013, 8, e75665. [Google Scholar] [CrossRef]
- Tomaras, G.D.; Ferrari, G.; Shen, X.; Alam, S.M.; Liao, H.X.; Pollara, J.; Bonsignori, M.; Moody, M.A.; Fong, Y.; Chen, X.; et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc. Natl. Acad. Sci. USA 2013, 110, 9019–9024. [Google Scholar] [CrossRef] [Green Version]
- Koblin, B.A.; Mayer, K.H.; Noonan, E.; Wang, C.Y.; Marmor, M.; Sanchez, J.; Brown, S.J.; Robertson, M.N.; Buchbinder, S.P. Sexual risk behaviors, circumcision status, and preexisting immunity to adenovirus type 5 among men who have sex with men participating in a randomized HIV-1 vaccine efficacy trial: Step study. J. Acquir. Immune Defic. Syndr. 2012, 60, 405–413. [Google Scholar] [CrossRef]
- Roff, S.R.; Noon-Song, E.N.; Yamamoto, J.K. The Significance of Interferon-γ in HIV-1 Pathogenesis, Therapy, and Prophylaxis. Front. Immunol. 2014, 4, 498. [Google Scholar] [CrossRef] [PubMed]
- Los Alamos National Laboratory (LANL), HIV Molecular Immunology Database: CTL/CD8+ Epitope Summary. Available online: https://www.hiv.lanl.gov/content/immunology/tables/ctl_summary.html (accessed on 30 October 2018).
- Vidya Vijayan, K.K.; Karthigeyan, K.P.; Tripathi, S.P.; Hanna, L.E. Pathophysiology of CD4+ T-cell depletion in HIV-1 and HIV-2 infections. Front. Immunol. 2017, 8, 580. [Google Scholar] [CrossRef] [PubMed]
- Simone, R.; Piatti, G.; Saverino, D. The inhibitory co-receptors: A way to save from anergy the HIV-specific T cells. Curr. HIV Res. 2009, 7, 266–272. [Google Scholar] [CrossRef]
- Altfeld, M.; Rosenberg, E.S.; Shankarappa, R.; Mukherjee, J.S.; Hecht, F.M.; Eldridge, R.L.; Addo, M.M.; Poon, S.H.; Phillips, M.N.; Robbins, G.K.; et al. Cellular immune responses and viral diversity in individuals treated during acute and early HIV-1 infection. J. Exp. Med. 2001, 193, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Sainski, A.M.; Natesampillai, S.; Bren, G.D.; Badley, A.D. Choice of antiretroviral therapy differentially impacts survival of HIV-infected CD4 T cells. Mol. Cell. Ther. 2014, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef]
- Rosás-Umbert, M.; Mothe, B.; Noguera-Julian, M.; Bellido, R.; Puertas, M.C.; Carrillo, J.; Rodriguez, C.; Perez-Alvarez, N.; Cobarsí, P.; Gomez, C.E.; et al. Virological and immunological outcome of treatment interruption in HIV-1-infected subjects vaccinated with MVA-B. PLoS ONE 2017, 12, e0184929. [Google Scholar] [CrossRef]
- Pinschewer, D.D. Virally vectored vaccine delivery: Medical needs, mechanisms, advantages and challenges. Swiss Med. Wkly. 2017, 147, w14465. [Google Scholar] [CrossRef]
- Huang, Y.; Duerr, A.; Frahm, N.; Zhang, L.; Moodie, Z.; De Rosa, S.; McElrath, M.J.; Gilbert, P.B. Immune-correlates analysis of an HIV-1 vaccine efficacy trial reveals an association of nonspecific interferon-γ secretion with increased HIV-1 infection risk: A cohort-based modeling study. PLoS ONE 2014, 9, e108631. [Google Scholar] [CrossRef]
- Rigby, M.A.; Mackay, N.; Reid, G.; Osborne, R.; Neil, J.C.; Jarrett, O. Immunogenicity of a peptide from a major neutralising determinant of the feline immunodeficiency virus surface glycoprotein. Vaccine 1996, 4, 1095–1102. [Google Scholar] [CrossRef]
- Lombardi, S.; Garzelli, C.; Pistello, M.; Massi, C.; Matteucci, D.; Baldinotti, F.; Cammarota, G.; da Prato, L.; Bandecchi, P.; Tozzini, F.; et al. A neutralizing antibody-inducing peptide of the V3 domain of feline immunodeficiency virus envelope glycoprotein does not induce protective immunity. J. Virol. 1994, 68, 8374–8379. [Google Scholar] [PubMed]
- Cerutti, N.; Loredo-Varela, J.L.; Caillat, C.; Weissenhorn, W. Antigp41 membrane proximal external region antibodies and the art of using the membrane for neutralization. Curr. Opin. HIV AIDS 2017, 12, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Planès, R.; Ben Haij, N.; Leghmari, K.; Serrero, M.; BenMohamed, L.; Bahraoui, E. HIV-1 Tat protein activates both the MyD88 and TRIF pathways to induce tumor necrosis factor alpha and interleukin-10 in human monocytes. J. Virol. 2016, 90, 5886–5898. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FIV or HIV Peptide ab | FIV or Counterpart HIV Protein (FIV/HIV Subtype) cd | Peptide AA No.cde | FIV or Counterpart HIV Peptide Sequence efg | FIV and HIV % AA Similarity (% AA Identity) [F/H Gap] dfg |
|---|---|---|---|---|
| Fp9-3 | p24 (A,B,C,D) | 15 | FAPARMQCRAWYLEA | 60% (13%) [H1] (44% (11%) [H1]) |
| Hp10-3 | p24 (B,A4,D,G,J,K) | 15 | NPPIPVGEIYKR-WII | 62% (12%) [H1] |
| MAP4 | p24 (A,B,C,D) | 15 | FAPARMQCRAWYLEA | 60% (13%) [H1] (44% (11%) [H1]) |
| Fp14-3 | p24 (A,B,D) | 14 | AEVKLYLKQSLSIA | 71% (29%) [0] (59% (24%) [0]) |
| Fp14-4 | p24 (A) | 13 | KLYLKQSLSIANA | 77% (31%) [0] (67% (27%) [0]) |
| Hp15-3/4 | p24 (B,A6,D,K) | 15 | VKNWMTETLLVQNAN | 80% (40%) [0] |
| MAP3 | p24 (A,B,D) | 16 | AEVKLYLKQSLSIANA | 75% (37%) [0] (65% (29%) [0]) |
| FRT3-3 | RT (B,D) | 15 | KKK-SGKWRMLIDFRV | 75% (69%) [F1] |
| FRT3-4 | RT (B,D) | 13 | WRMLIDFRVLNKL | 77% (69%) [0] (71% (64%) [0]) |
| HRT3-3 | RT (B,A,C,D,F,G,H,J,K) | 15 | KKKDSTKWRKLVDFR | 75% (69%) [F1] |
| HRT3-4 | RT (B A,C,D,F,G,H,J,K) | 14 | KWRKLVDFRELNKR | 79% (71%) [0] |
| MAP2 | RT (B,D) h | 18 | KKK-SGKWR-LIDFRVLNKL | 75% (70%) [F2] |
| MAP2v | RT (B,D) h | 19 | KKK-SGKWRLLIDFRVLNKL | 75% (70%) [F1] |
| FRT7-1 | RT (B,C,D) | 15 | GRRYVWCSLPQGWVL | 60% (53%) [0] |
| FRT7-2 | RT (B,C,D) | 14 | CSLPQGWVLSPLIY | 64% (57%) [0] |
| HRT7-1 | RT (B,Ai,C,D,F,G,H,J) | 15 | GIRYQYNVLPQGWKG | 60% (53%) [0] |
| HRT7-2 | RT (B,A,C,D,F,G,H,J,K) | 14 | NVLPQGWKGSPAIF | 64% (57%) [0] |
| MAP5 | RT (B,C,D) | 20 | GRRYVWCSLPQGWVLSPLIY | 65% (55%) [0] |
| FIV Peptide | % Responder Rate of Vaccinated Cats a | Percent Responder Rate of HIV+ Human Subjects a | In Silico HLA Analysis b | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Proliferation acd | Cytokine ac | FIV | Proliferation acd | Perforin/GrzA/GrzB/CD107a cd | Cytokine a | NetMHC 4.0 | NetMHC 2.3 | |||||||
| CD4 T | CD8 T | IFNγ | IL-2 | E/S e | CD4 T | CD8 T | CD4 CTLa | RF f | CD8 CTLa | RF f | IFNγ | HLA-ABC g | DRB1 | |
| Fp9-3 | 66.7 | 66.7 | 66.7 | 0.0 | S | 27.8 | 88.9 | 60.0 | 0.389 h | 70.0 | 0.444 h | 9.1 | A2, B8, B44 B27, C07, B83, C06 | - i |
| Fp14-3 | 33.3 | 33.3 | 33.3 | 33.3 | - | 0.0 | 83.3 | 70.0 | 0.444 h | 70.0 | 0.389 h | 66.7 | A2, A24, A1, B44, C14 | 1201, 0405, 1302, 0404, 1602, 0401, 1001, 0101 |
| Fp14-4 | 33.3 | 33.3 | 33.3 | 16.7 | - | 16.7 | 100.0 | 100.0 | 0.444 h | 100.0 | 0.444 h | 33.3 | A2, A24, A1 | 0701, 1001, 1201, 0405, 1302, 0404, 0401, 1602, 0101 |
| FRT3-3 | 42.9 | 42.9 | 14.3 | 14.3 | E | 0.0 | 62.5 | 66.7 | 0.444 | 83.3 | 0.278 | 100.0 | B27, C15, A3, A1 | 1602, 1001, 0405, 0101, 1501 |
| FRT3-4 | 42.9 | 57.1 | 28.6 | 42.9 | S | 0.0 | 37.5 | 66.7 | 0.389 | 66.7 | 0.444 | 10.0 | B27, A1, B44, A3 | 0301, 1602, 0101, 0103, 0401, 0405, 1001,1501 |
| FRT7-1 | 100.0 | 100.0 | 33.3 | 66.7 | S j | 20.0 | 20.0 | 60.0 | 0.467 | 100.0 | 0.667 | 0.0 | B27, A1, C07, A24, C06, C07, C08 | 0404 |
| FRT7-2 | 66.7 | 66.7 | 33.3 | 0.0 | S j | 0.0 | 0.0 | 100.0 | 0.533 | 100.0 | 0.867 | 0.0 | A1, B27, A29, C15, A3, C08 | 1201 |
| Average | 55.1 | 57.1 | 34.7 | 24.8 | 6.4 | 56.4 | 74.8 | 0.444 | 84.3 | 0.505 | 31.3 | |||
| p-value k | 0.8768 | 0.3898 | 0.0129 | 0.3060 | 0.4583 | |||||||||
| Group | Immunization ab | No. of Protected/Total No. of Cats a [% Protection] | Preventable | p-value ad | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. a | Vaccine 1 | Vaccine 2 | Vaccine 3 | Route | 0wpc | 3wpc | 6wpc | 9wpc | 12wpc | 16-24wpc | Fraction c (%) | 1abc vs. 2a–d |
| Individual Groups versus Combined Group 2a–d | ||||||||||||
| 1a | MAP2/3/4t | MAP2/2v/3/5 | MAP2/2v/3/5 | SC/ID | 6/6 | 6/6 | 4/6 | 4/6 | 3/6 | 3/6 | 20% | 0.6550 |
| 1b | MAP2v/3/4/5 | MAP2v/3/4/5 | MAP2v/3/4/5 | SC | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 6/6 | 100% | 0.0152 |
| 1c | MAP2v/3/4/5 | MAP2v/3/4/5 | MAP2v/3/4/5 | ID | 7/7 | 7/7 | 6/7 | 6/7 | 6/7 | 6/7 | 77.1% | 0.0686 |
| 2a-d | Adj,PBS,- | Adj,PBS,- | Adj,PBS,- | SC/ID, SC, ID, - | 16/16 | 15/16 | 8/16 | 6/16 | 6/16 | 6/16 | - | - |
| 2a | Adj | Adj | Adj | SC/ID | 3/3 | 2/3 | 1/3 | 1/3 | 1/3 | 1/3 | - | - |
| 2b | PBS | PBS | PBS | SC/ID | 5/5 | 5/5 | 4/5 | 3/5 | 3/5 | 3/5 | - | - |
| 2c | Adj | Adj | Adj | SC, ID | 6/6 | 6/6 | 2/6 | 2/6 | 2/6 | 2/6 | - | - |
| 2d | - | - | - | - | 2/2 | 2/2 | 1/2 | 0/2 | 0/2 | 0/2 | - | - |
| Combined Groups 1abc or 1bc versus Combined Group 2a–d | ||||||||||||
| 1abc | MAP2/3/4t MAP2v/3/4/5 | MAP2/2v/3/5 MAP2v/3/4/5 | MAP2/2v/3/5 MAP2v/3/4/5 | SC/ID, SC, ID | 19/19[100%] | 19/19[100%] | 16/19[84%] | 16/19[84%] | 15/19[79%] | 15/19[79%] | 66.3% | 0.0180 |
| 1bc | MAP2v/3/4/5 | MAP2v/3/4/5 | MAP2v/3/4/5 | SC, ID | 13/13 | 13/13 | 12/13 | 12/13 | 12/13 | 12/13 | 87.7% | 0.0057 |
| Unprotected versus | 3-6w Post-2nd Vac a | 3w Post-3rd Vac a | 6wk Post-3rd Vac a | 3-6w Post-2nd Vac a | 3w Post-3rd Vac a | 6w Post-3rd Vac a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Protected (Table No.) b | CD4 T cd | CD8 T cd | CD4 T cd | CD8 T cd | CD4 T cd | CD8 T cd | IL-2 cd | IFNγ c | IL-2 cd | IFNγ cd | IL-2 cd | IFNγ cd |
| Percent responder rate e | ||||||||||||
| Group-1 unprotected (S5 vs. S6) | 18.2 | 13.6 | 18.2 | 22.7 | 47.2 h | 41.1 h | 27.3 | 47.7 | 18.2 | 61.4 | 11.4 | 31.8 |
| Group-1bc protected (S5 vs. S6) | 40.6 | 27.3 | 65.7 | 55.9 | 67.1 h | 59.4 h | 41.3 | 38.5 | 24.5 | 27.3 | 21.7 | 26.6 |
| p-valueg (S5 vs. S6) | 0.0078 | 0.0648 | 0.00003 | 0.0018 | 0.2308 | 0.1353 | 0.2821 | 0.4747 | 0.4599 | 0.0173 | 0.2589 | 0.6396 |
| Group-1 unprotected (5) | 18.2 | 15.9 | 18.2 | 22.8 | 48.0 i | 40.2 i | 27.3 | 47.8 | 18.2 | 61.3 | 11.4 | 31.8 |
| Group-1 protected (5) | 39.2 | 30.1 | 59.1 | 55.1 | 67.7 i | 56.6 i | 38.6 | 38.1 | 22.7 | 25.6 | 18.2 | 24.4 |
| p-valueg (5) | 0.0732 | 0.0335 | 0.0038 | 0.0004 | 0.0125 | 0.3079 | 0.1374 | 0.2754 | 0.6435 | 0.0041 | 0.4511 | 0.6441 |
| Average magnitude of the positive responses f | ||||||||||||
| Group-1 unprotected (S5 vs. S6) | 2.8 | 7.6 | 2.6 | 14.8 | 3.2 h | 5.6 h | 70.6 | 181.4 | 53.6 | 228.4 | 34.6 | 127.9 |
| Group-1bc protected (S5 vs. S6) | 5.8 | 7.5 | 10.6 | 9.0 | 10.8 h | 7.6 h | 125 | 142.3 | 153.6 | 240.3 | 175 | 172.3 |
| p-valueg (S5 vs. S6) | 0.1014 | 0.9992 | 0.0083 | 0.5740 | 0.0471 | 0.5698 | 0.0781 | 0.3902 | 0.0266 | 0.8993 | 0.0072 | 0.4951 |
| Group-1 unprotected (5) | 3.4 | 22.5 | 2.8 | 27.8 | 5.3 i | 6.7 i | 158.3 | 254.5 | 68.6 | 262.2 | 54.0 | 117.4 |
| Group-1 protected (5) | 4.5 | 14.2 | 9.3 | 8.2 | 9.8i | 8.1 i | 137.7 | 159.7 | 112.1 | 156.2 | 129.6 | 145.5 |
| p-valueg (5) | 0.6626 | 0.4710 | 0.0022 | 0.3311 | 0.0529 | 0.5551 | 0.5263 | 0.2959 | 0.3436 | 0.2230 | 0.1257 | 0.6801 |
| Protected/Vaccinated Cats (P) | Unprotected Cats (U) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immune | Vaccine Group 1a a | Vaccine Group 1b a | Vaccine Group 1c (G1c) a | Vaccine Group 1a a | G1cab | U vs. P b | |||||||||||||||
| Anal. b | BFA | DU4 | DX5 | HOC | RL2 | RN4 | RO1 | RO7 | RP4 | RQ1 | HOB | RL4 | RN3 | RO2 | RP3 | RQ5 | DU5 | DX1 | OLL | RL1 | p-value c |
| Percent responder rated | |||||||||||||||||||||
| CD8 T | 44.4 f | 33.3 f | 55.6 f | 72.7 | 81.8 | 63.6 | 63.6 | 100.0 | 100.0 | 63.6 | 54.5 | 0.0 | 18.2 | 36.4 | 45.5 | 72.7 | 11.1 f | 55.6 f | 66.7 f | 27.3 | 0.3079 |
| CD4 T | 77.8 f | 55.6 f | 77.8 f | 63.6 | 36.4 | 72.7 | 81.8 | 90.9 | 81.8 | 90.9 | 54.5 | 54.5 | 63.6 | 45.5 | 81.8 | 54.5 | 55.6 f | 55.6 f | 44.4 f | 36.4 | 0.0125 |
| Average | 61.1 | 44.5 | 66.7 | 68.2 | 59.1 | 68.2 | 72.7 | 95.5 | 90.9 | 77.3 | 54.5 | 27.3 | 40.9 | 41.0 | 63.7 | 63.6 | 33.4 | 55.6 | 55.6 | 31.9 | |
| IL-2 | 9.1 | 0.0 | 0.0 | 0.0 | 9.1 | 54.5 | 18.2 | 9.1 | 18.2 | 54.5 | 0.0 | 0.0 | 36.4 | 0.0 | 36.4 | 45.5 | 0.0 | 0.0 | 18.2 | 27.3 | 0.4511 |
| IFNγ | 36.4 | 0.0 | 9.1 | 0.0 | 0.0 | 36.4 | 36.4 | 9.1 | 0.0 | 9.1 | 63.6 | 0.0 | 54.5 | 45.5 | 36.4 | 54.5 | 0.0 | 18.2 | 54.5 | 54.5 | 0.6441 |
| Average | 22.8 | 0.0 | 4.6 | 0.0 | 4.6 | 45.5 | 27.3 | 9.1 | 9.1 | 31.8 | 31.8 | 0.0 | 45.5 | 22.8 | 36.4 | 50.0 | 0.0 | 9.1 | 36.4 | 40.9 | |
| Average magnitude of positive responsese | |||||||||||||||||||||
| CD8 T | 21.2 | 6.7 | 6.2 | 6.9 | 3.1 | 9.5 | 2.6 | 9.6 | 9.3 | 6.6 | 6.2 | 0.0 | 11.3 | 3.7 | 10.4 | 16.2 | 2.0 | 9.2 | 9.5 | 6.2 | 0.5551 |
| CD4 T | 3.9 | 1.6 | 2.3 | 14.4 | 7.9 | 11.7 | 10.9 | 10.3 | 10.9 | 20.1 | 7.0 | 4.1 | 11.6 | 4.8 | 15.6 | 20.1 | 9.2 | 3.3 | 2.7 | 5.9 | 0.0529 |
| Average | 12.5 | 4.1 | 4.2 | 10.7 | 5.5 | 10.6 | 6.7 | 10.0 | 10.1 | 13.3 | 6.6 | 2.1 | 11.4 | 4.3 | 13.0 | 18.1 | 5.6 | 6.2 | 6.1 | 6.0 | |
| IL-2 | 51 | 0 | 0 | 0 | 372 | 205 | 373 | 105 | 238 | 214 | 0 | 0 | 144 | 0 | 255 | 119 | 0 | 0 | 105 | 111 | 0.1257 |
| IFNγ | 78 | 0 | 56 | 0 | 0 | 96 | 133 | 116 | 0 | 546 | 382 | 0 | 237 | 179 | 362 | 144 | 0 | 63 | 192 | 215 | 0.6801 |
| Average | 33 | 0 | 28 | 0 | 186 | 150 | 253 | 111 | 119 | 380 | 191 | 0 | 190 | 90 | 309 | 132 | 0 | 31 | 148 | 163 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahay, B.; Aranyos, A.M.; Mishra, M.; McAvoy, A.C.; Martin, M.M.; Pu, R.; Shiomitsu, S.; Shiomitsu, K.; Dark, M.J.; Sanou, M.P.; et al. Immunogenicity and Efficacy of a Novel Multi-Antigenic Peptide Vaccine Based on Cross-Reactivity between Feline and Human Immunodeficiency Viruses. Viruses 2019, 11, 136. https://doi.org/10.3390/v11020136
Sahay B, Aranyos AM, Mishra M, McAvoy AC, Martin MM, Pu R, Shiomitsu S, Shiomitsu K, Dark MJ, Sanou MP, et al. Immunogenicity and Efficacy of a Novel Multi-Antigenic Peptide Vaccine Based on Cross-Reactivity between Feline and Human Immunodeficiency Viruses. Viruses. 2019; 11(2):136. https://doi.org/10.3390/v11020136
Chicago/Turabian StyleSahay, Bikash, Alek M. Aranyos, Meerambika Mishra, Andrew C. McAvoy, Marcus M. Martin, Riuyu Pu, Sayaka Shiomitsu, Keijiro Shiomitsu, Michael J. Dark, Missa P. Sanou, and et al. 2019. "Immunogenicity and Efficacy of a Novel Multi-Antigenic Peptide Vaccine Based on Cross-Reactivity between Feline and Human Immunodeficiency Viruses" Viruses 11, no. 2: 136. https://doi.org/10.3390/v11020136