Metagenomic Characterization of the Viral Community of the South Scotia Ridge

 , and
, and
Abstract
1. Introduction
2. Methods and Materials
2.1. Sample Collection and Sequencing
2.2. Virome Composition Analysis
2.3. Virome Comparison Analysis
2.4. Metagenomic Assembly and Function Analysis
2.5. Phylogenetic Analysis
2.6. Genomic Comparison
3. Results
3.1. Overview of SSR Viromes
3.2. Taxonomic Diversity Analysis
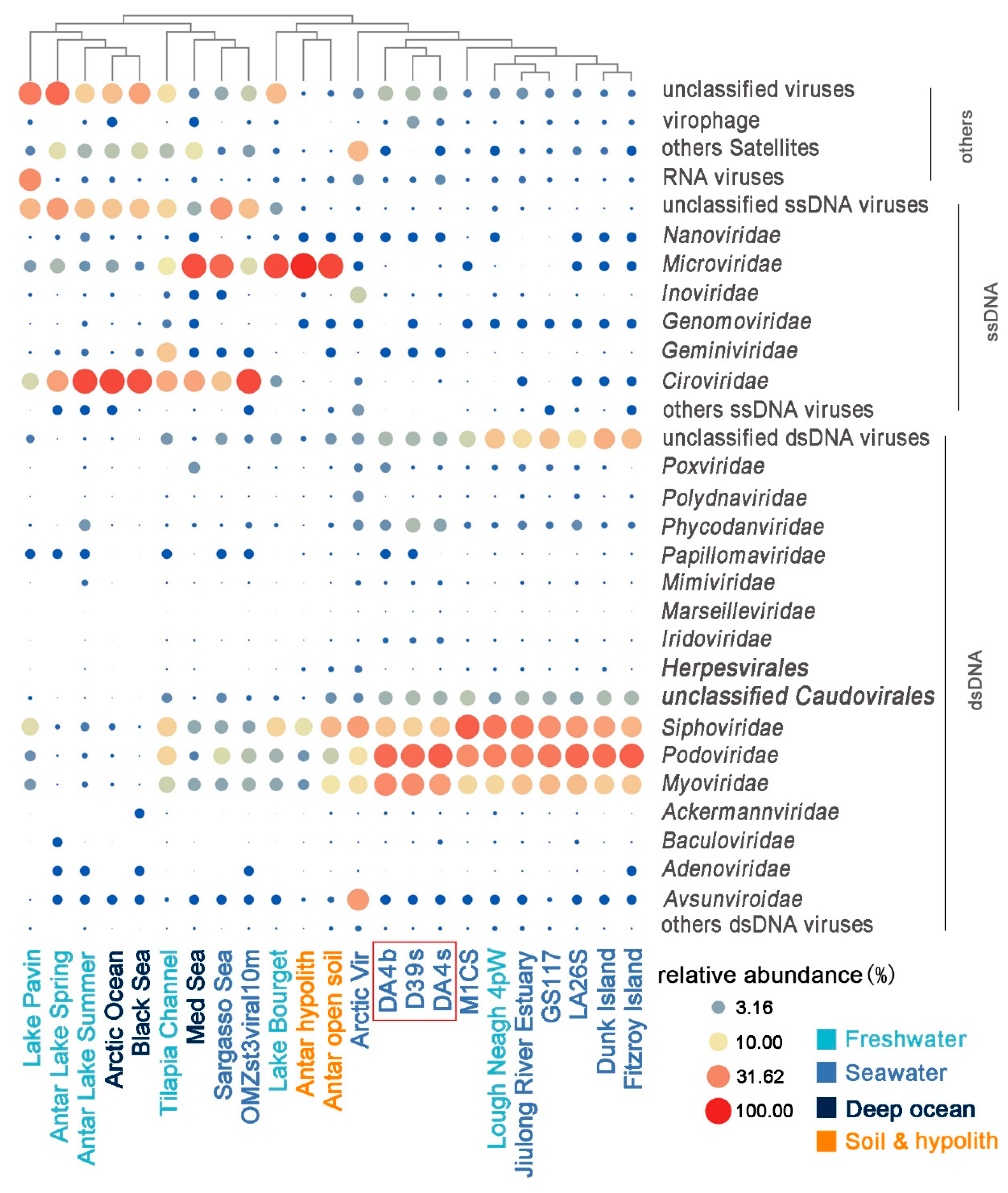
3.3. Comparison with Other Viromes
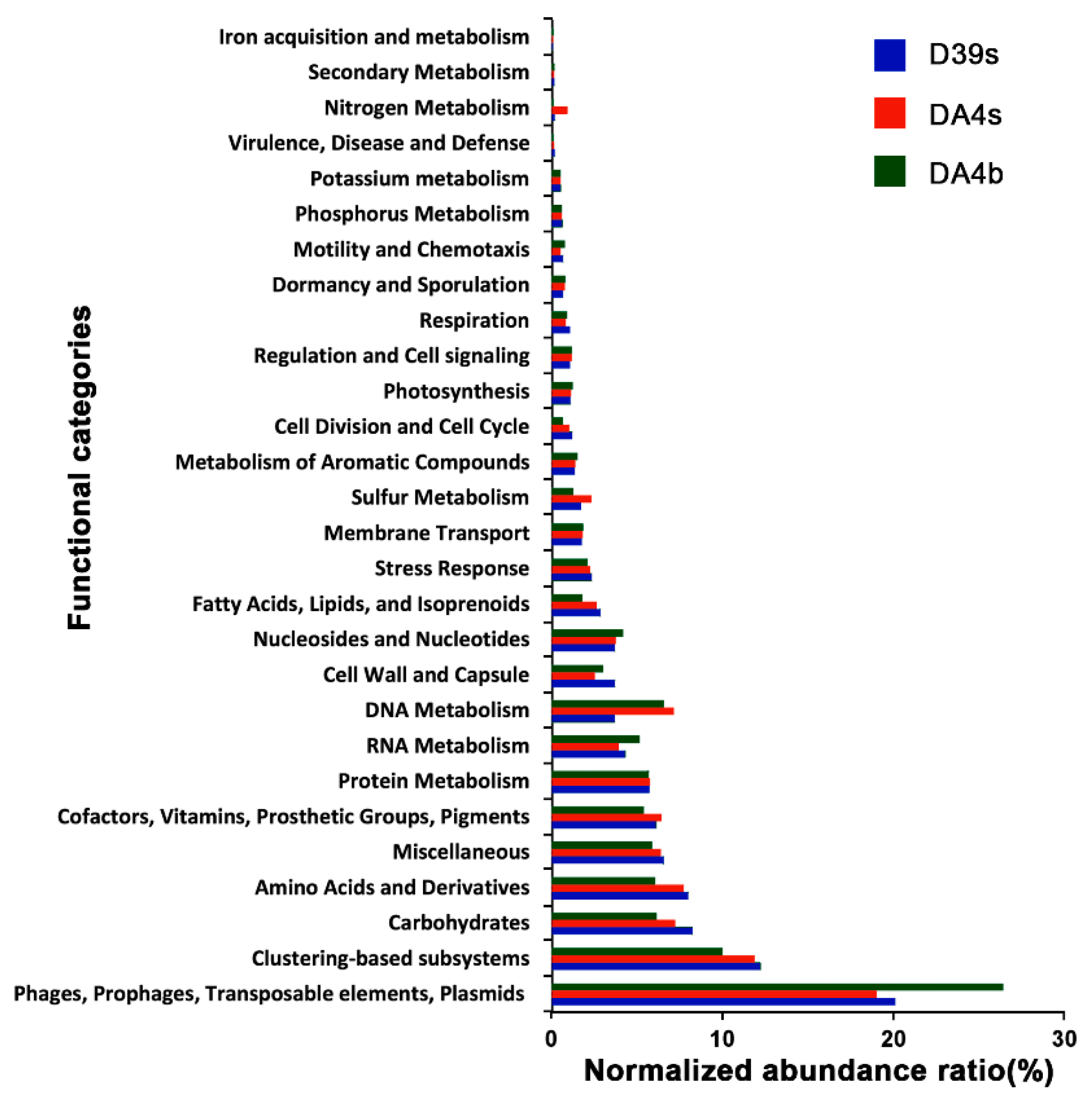
3.4. Contigs and Function Analysis
3.5. Phylogenetic Tree Analysis
3.5.1. Terminase Phylogeny
3.5.2. Capsid_NCLDV Phylogeny
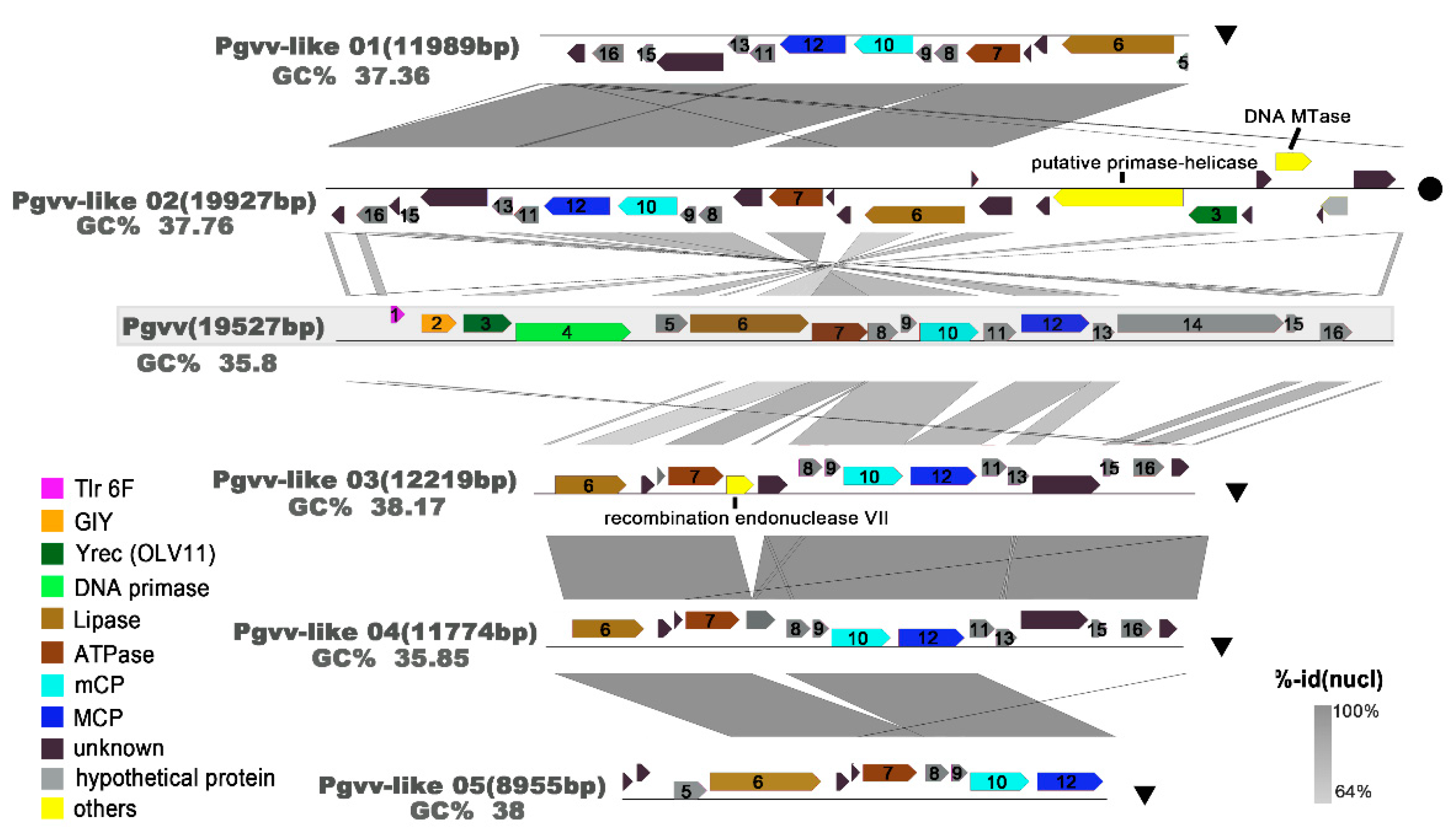
3.6. Novel Pgvv Group
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rohwer, F. Global phage diversity. Cell 2003, 113, 141. [Google Scholar] [CrossRef]
- Lipkin, W.I. Microbe hunting. Microbiol. Mol. Biol. Rev. 2010, 74, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Proctor, L.M.; Fuhrman, J.A. Viral mortality of marine-bacteria and cyanobacteria. Nature 1990, 343, 60–62. [Google Scholar] [CrossRef]
- Suttle, C.A. The significance of viruses to mortality in aquatic microbial communities. Microb. Ecol. 1994, 28, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.D.; Wilhelm, S.W.; Thingstad, F.; Weinbauer, M.G.; Bratbak, G.; Heldal, M.; Kimmance, S.A.; Middelboe, M.; Nagasaki, K.; Paul, J.H.; et al. Global-scale processes with a nanoscale drive: The role of marine viruses. ISME J. 2008, 2, 575–578. [Google Scholar] [CrossRef]
- Rohwer, F.; Thurber, R.V. Viruses manipulate the marine environment. Nature 2009, 459, 207–212. [Google Scholar] [CrossRef]
- Weitz, J.S.; Wilhelm, S.W. Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000 Biol. Rep. 2012, 4, 17. [Google Scholar] [CrossRef]
- Jover, L.F.; Effler, T.C.; Buchan, A.; Wilhelm, S.W.; Weitz, J.S. The elemental composition of virus particles: Implications for marine biogeochemical cycles. Nat. Rev. Microbiol. 2014, 12, 519–528. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.H.; Cook, A.; Millard, A.; Bailey, S.; Clokie, M. Marine ecosystems: Bacterial photosynthesis genes in a virus. Nature 2003, 424, 741. [Google Scholar] [CrossRef] [PubMed]
- Kuparinen, J.; Bjornsen, P.K. Spatial-distribution of bacterioplankton production across the weddell-scotia confluence during early austral summer 1988–1989. Pol. Biol. 1992, 12, 197–204. [Google Scholar] [CrossRef]
- Archer, S.D.; Leakey, R.J.G.; Burkill, P.H.; Sleigh, M.A. Microbial dynamics in coastal waters of east antarctica: Herbivory by heterotrophic dinoflagellates. Mar. Ecol. Prog. Ser. 1996, 139, 239–255. [Google Scholar] [CrossRef]
- Vaque, D.; Calderon-Paz, J.I.; Guixa-Boixereu, N.; Pedros-Alio, C. Spatial distribution of microbial biomass and activity (bacterivory and bacterial production) in the northern weddell sea during the austral summer (January 1994). Aquat. Microb. Ecol. 2002, 29, 107–121. [Google Scholar] [CrossRef]
- Christaki, U.; Obernosterer, I.; Van Wambeke, F.; Veldhuis, M.; Garcia, N.; Catala, P. Microbial food web structure in a naturally iron-fertilized area in the southern ocean (kerguelen plateau). Deep-Sea Res. Part II 2008, 55, 706–719. [Google Scholar] [CrossRef]
- Montes-Hugo, M.; Doney, S.C.; Ducklow, H.W.; Fraser, W.; Martinson, D.; Stammerjohn, S.E.; Schofield, O. Recent changes in phytoplankton communities associated with rapid regional climate change along the western antarctic peninsula. Science 2009, 323, 1470–1473. [Google Scholar] [CrossRef]
- Moschonas, G.; Gowen, R.J.; Paterson, R.F.; Mitchell, E.; Stewart, B.M.; McNeill, S.; Glibert, P.M.; Davidson, K. Nitrogen dynamics and phytoplankton community structure: The role of organic nutrients. Biogeochemistry 2017, 134, 125–145. [Google Scholar] [CrossRef]
- Guixa-Boixereu, N.; Vaque, D.; Gasol, J.M.; Sanchez-Camara, J.; Pedros-Alio, C. Viral distribution and activity in antarctic waters. Deep-Sea Res. Part II 2002, 49, 827–845. [Google Scholar] [CrossRef]
- Yau, S.; Lauro, F.M.; DeMaere, M.Z.; Brown, M.V.; Thomas, T.; Raftery, M.J.; Andrews-Pfannkoch, C.; Lewis, M.; Hoffman, J.M.; Gibson, J.A.; et al. Virophage control of antarctic algal host-virus dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 6163–6168. [Google Scholar] [CrossRef]
- Malits, A.; Christaki, U.; Obernosterer, I.; Weinbauer, M.G. Enhanced viral production and virus-mediated mortality of bacterioplankton in a natural iron-fertilized bloom event above the kerguelen plateau. Biogeosciences 2014, 11, 6841–6853. [Google Scholar] [CrossRef]
- Vaque, D.; Boras, J.A.; Torrent-Llagostera, F.; Agusti, S.; Arrieta, J.M.; Lara, E.; Castillo, Y.M.; Duarte, C.M.; Sala, M.M. Viruses and protists induced-mortality of prokaryotes around the antarctic peninsula during the austral summer. Front. Microbiol. 2017, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bueno, A.; Tamames, J.; Velazquez, D.; Moya, A.; Quesada, A.; Alcami, A. High diversity of the viral community from an antarctic lake. Science 2009, 326, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Zawar-Reza, P.; Arguello-Astorga, G.R.; Kraberger, S.; Julian, L.; Stainton, D.; Broady, P.A.; Varsani, A. Diverse small circular single-stranded DNA viruses identified in a freshwater pond on the mcmurdo ice shelf (antarctica). Infect. Genet. Evol. 2014, 26, 132–138. [Google Scholar] [CrossRef] [PubMed]
- De Carcer, D.A.; Lopez-Bueno, A.; Pearce, D.A.; Alcami, A. Biodiversity and distribution of polar freshwater DNA viruses. Sci Adv. 2015, 1, e1400127. [Google Scholar] [CrossRef] [PubMed]
- De Carcer, D.A.; Lopez-Bueno, A.; Alonso-Lobo, J.M.; Quesada, A.; Alcami, A. Metagenomic analysis of lacustrine viral diversity along a latitudinal transect of the antarctic peninsula. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar]
- Cavicchioli, R.; Erdmann, S. The discovery of antarctic rna viruses: A new game changer. Mol. Ecol. 2015, 24, 4809–4811. [Google Scholar] [CrossRef]
- Brum, J.R.; Hurwitz, B.L.; Schofield, O.; Ducklow, H.W.; Sullivan, M.B. Seasonal time bombs: Dominant temperate viruses affect southern ocean microbial dynamics. ISME J. 2016, 10, 437–449. [Google Scholar] [CrossRef]
- Hopkins, M.; Kailasan, S.; Cohen, A.; Roux, S.; Tucker, K.P.; Shevenell, A.; Agbandje-McKenna, M.; Breitbart, M. Diversity of environmental single-stranded DNA phages revealed by pcr amplification of the partial major capsid protein. ISME J. 2014, 8, 2093–2103. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Kramer, R.; Van Goethem, M.W.; Makhalanyane, T.P.; Hogg, I.; Cowan, D.A. Environmental drivers of viral community composition in antarctic soils identified by viromics. Microbiome 2017, 5, 83. [Google Scholar] [CrossRef]
- Howe, J.A.; Livermore, R.A.; Maldonado, A. Mudwave activity and current-controlled sedimentation in powell basin, northern weddell sea, antarctica. Mar. Geol. 1998, 149, 229–241. [Google Scholar] [CrossRef]
- Sun, G.; Xiao, J.; Wang, H.; Gong, C.; Pan, Y.; Yan, S.; Wang, Y. Efficient purification and concentration of viruses from a large body of high turbidity seawater. MethodsX 2014, 1, 197–206. [Google Scholar] [CrossRef] [PubMed]
- SRA. Available online: https://www.ncbi.nlm.nih.gov/sra/PRJNA505984 (accessed on 20 November 2018).
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Download data from NCBI FTP site. Available online: ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/ (accessed on 18 July 2018).
- Download data from NCBI FTP site. Available online: ftp://ftp.ncbi.nlm.nih.gov/refseq/release/viral/ (accessed on 18 July 2018).
- Roux, S.; Faubladier, M.; Mahul, A.; Paulhe, N.; Bernard, A.; Debroas, D.; Enault, F. Metavir: A web server dedicated to virome analysis. Bioinformatics 2011, 27, 3074–3075. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Robin, A.; Ravet, V.; Personnic, S.; Theil, S.; Colombet, J.; Sime-Ngando, T.; Debroas, D. Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS ONE 2012, 7, e33641. [Google Scholar] [CrossRef] [PubMed]
- Skvortsov, T.; de Leeuwe, C.; Quinn, J.P.; McGrath, J.W.; Allen, C.C.R.; McElarney, Y.; Watson, C.; Arkhipova, K.; Lavigne, R.; Kulakov, L.A. Metagenomic characterisation of the viral community of lough neagh, the largest freshwater lake in Ireland. PLoS ONE 2016, 11, e0150361. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Brito, B.; Li, L.L.; Wegley, L.; Furlan, M.; Angly, F.; Breitbart, M.; Buchanan, J.; Desnues, C.; Dinsdale, E.; Edwards, R.; et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010, 4, 739–751. [Google Scholar] [CrossRef]
- Cassman, N.; Prieto-Davo, A.; Walsh, K.; Silva, G.G.Z.; Angly, F.; Akhter, S.; Barott, K.; Busch, J.; McDole, T.; Haggerty, J.M.; et al. Oxygen minimum zones harbour novel viral communities with low diversity. Environ. Microbiol. 2012, 14, 3043–3065. [Google Scholar] [CrossRef]
- Williamson, S.J.; Allen, L.Z.; Lorenzi, H.A.; Fadrosh, D.W.; Brami, D.; Thiagarajan, M.; McCrow, J.P.; Tovchigrechko, A.; Yooseph, S.; Venter, J.C. Metagenomic exploration of viruses throughout the Indian ocean. PLoS ONE 2012, 7, e42047. [Google Scholar] [CrossRef]
- Cai, L.L.; Zhang, R.; He, Y.; Feng, X.Y.; Jiao, N.Z. Metagenomic analysis of virioplankton of the subtropical jiulong river estuary, China. Viruses 2016, 8, 35. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; Sullivan, M.B. The pacific ocean virome (POV): A marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS ONE 2013, 8, e57355. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol. 2006, 4, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Dell’Anno, A.; Corinaldesi, C.; Magagnini, M.; Noble, R.; Tamburini, C.; Weinbauer, M. Major viral impact on the functioning of benthic deep-sea ecosystems. Nature 2008, 454, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Zablocki, O.; van Zyl, L.; Adriaenssens, E.M.; Rubagotti, E.; Tuffin, M.; Cary, S.C.; Cowan, D. High-level diversity of tailed phages, eukaryote-associated viruses, and virophage-like elements in the metaviromes of antarctic soils. Appl. Environ. Microbiol. 2014, 80, 6888–6897. [Google Scholar] [CrossRef]
- Dixon, P. Vegan, a package of r functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Coutinho, F.H.; Silveira, C.B.; Gregoracci, G.B.; Thompson, C.C.; Edwards, R.A.; Brussaard, C.P.D.; Dutilh, B.E.; Thompson, F.L. Marine viruses discovered via metagenomics shed light on viral strategies throughout the oceans. Nat. Commun. 2017, 8, 15955. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, N.; Pop, M. Sequence assembly demystified. Nat. Rev. Genet. 2013, 14, 157–167. [Google Scholar] [CrossRef] [PubMed]
- De Crcer, D.A.; Angly, F.E.; Alcami, A. Evaluation of viral genome assembly and diversity estimation in deep metagenomes. Bmc Genomics 2014, 15, 989. [Google Scholar]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.M.A.; Markowitz, V.M.; Chu, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.H.; Andersen, E.; Huntemann, M.; et al. Img/m: Integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017, 45, D507–D516. [Google Scholar] [CrossRef]
- DOE Joint Genome Insitute-IMG. Available online: https://img.jgi.doe.gov/ (accessed on 23 August 2018).
- Huntemann, M.; Ivanova, N.N.; Mavromatis, K.; Tripp, H.J.; Paez-Espino, D.; Tennessen, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Chen, I.M.A.; et al. The standard operating procedure of the DOE-JGI Metagenome Annotation Pipeline (MAP v.4). Stand. Genomic Sci. 2016, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics rast server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- MG-RAST metagenomics analysis server. Available online: http://metagenomics.anl.gov/ (accessed on 9 September 2018).
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (ITOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Yutin, N.; Shevchenko, S.; Kapitonov, V.; Krupovic, M.; Koonin, E.V. A novel group of diverse polinton-like viruses discovered by metagenome analysis. BMC Biol. 2015, 13, 95. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a web browser. BMC Bioinformatics 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Oh, H.M.; Kang, D.; Cho, J.C. Genome of a sar116 bacteriophage shows the prevalence of this phage type in the oceans. Proc. Natl. Acad. Sci. USA 2013, 110, 12343–12348. [Google Scholar] [CrossRef]
- Eggleston, E.M.; Hewson, I. Abundance of two pelagibacter ubique bacteriophage genotypes along a latitudinal transect in the north and south atlantic oceans. Front. Microbiol. 2016, 7, 1534. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef]
- Brum, J.R.; Sullivan, M.B. Rising to the challenge: Accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 2015, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Millard, A.D.; Pearce, D.; Zwirglmaier, K. Biogeography of bacteriophages at four hydrothermal vent sites in the antarctic based on g23 sequence diversity. FEMS Microbiol. Lett. 2016, 363, fnw043. [Google Scholar] [CrossRef] [PubMed]
- Flaviani, F.; Schroeder, D.C.; Lebret, K.; Balestreri, C.; Highfield, A.C.; Schroeder, J.L.; Thorpe, S.E.; Moore, K.; Pasckiewicz, K.; Pfaff, M.C.; et al. Distinct oceanic microbiomes from viruses to protists located near the antarctic circumpolar current. Front. Microbiol. 2018, 9, 1474. [Google Scholar] [CrossRef]
- Fancello, L.; Trape, S.; Robert, C.; Boyer, M.; Popgeorgiev, N.; Raoult, D.; Desnues, C. Viruses in the desert: A metagenomic survey of viral communities in four perennial ponds of the mauritanian sahara. ISME J. 2013, 7, 359–369. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; Deng, L.; Poulos, B.T.; Sullivan, M.B. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ. Microbiol. 2013, 15, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Beumer, A.; Robinson, J.B. A broad-host-range, generalized transducing phage (sn-t) acquires 16s rRNA genes from different genera of bacteria. Appl. Environ. Microb. 2005, 71, 8301–8304. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Roy, K.; Williamson, K.E.; White, D.C.; Wommack, K.E.; Sublette, K.L.; Radosevich, M. Prevalence of lysogeny among soil bacteria and presence of 16s rRNA and tRZN genes in viral-community DNA. Appl. Environ. Microb. 2008, 74, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Ravet, V.; Colombet, J.; Bettarel, Y.; Auguet, J.C.; Bouvier, T.; Lucas-Staat, S.; Vellet, A.; Prangishvili, D.; et al. Analysis of metagenomic data reveals common features of halophilic viral communities across continents. Environ. Microbiol. 2016, 18, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.L.; Temperton, B.; Thrash, J.C.; Schwalbach, M.S.; Vergin, K.L.; Landry, Z.C.; Ellisman, M.; Deerinck, T.; Sullivan, M.B.; Giovannoni, S.J. Abundant SAR11 viruses in the ocean. Nature 2013, 494, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Howard, E.C.; Sun, S.L.; Biers, E.J.; Moran, M.A. Abundant and diverse bacteria involved in dmsp degradation in marine surface waters. Environ. Microbiol. 2008, 10, 2397–2410. [Google Scholar] [CrossRef]
- Choi, D.H.; Park, K.T.; An, S.M.; Lee, K.; Cho, J.C.; Lee, J.H.; Kim, D.; Jeon, D.; Noh, J.H. Pyrosequencing revealed SAR116 clade as dominant dddp-containing bacteria in oligotrophic nw pacific ocean. PLoS ONE 2015, 10, e0116271. [Google Scholar] [CrossRef]
- Baudoux, A.C.; Brussaard, C.P.D. Characterization of different viruses infecting the marine harmful algal bloom species phaeocystis globosa. Virology 2005, 341, 80–90. [Google Scholar] [CrossRef]
- DiTullio, G.R.; Grebmeier, J.M.; Arrigo, K.R.; Lizotte, M.P.; Robinson, D.H.; Leventer, A.; Barry, J.B.; VanWoert, M.L.; Dunbar, R.B. Rapid and early export of phaeocystis antarctica blooms in the ross sea, antarctica. Nature 2000, 404, 595–598. [Google Scholar] [CrossRef]
- Lasken, R.S.; Stockwell, T.B. Mechanism of chimera formation during the multiple displacement amplification reaction. BMC Biotechnol. 2007, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Bae, J.W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microb. 2011, 77, 7663–7668. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Schenck, R.O.; Sullivan, M.B. Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 2013, 7, 1738–1751. [Google Scholar] [CrossRef] [PubMed]
- Maniloff, J.; Ackermann, H.W. Taxonomy of bacterial viruses: Establishment of tailed virus genera and the order caudovirales. Arch. Virol. 1998, 143, 2051–2063. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.M.; Rodriguez-Valera, F.; Garcia-Heredia, I.; Martin-Cuadrado, A.B.; Ghai, R. Reconstruction of novel cyanobacterial siphovirus genomes from mediterranean metagenomic fosmids. Appl. Environ. Microb. 2013, 79, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Tidona, C.A.; Schnitzler, P.; Kehm, R.; Darai, G. Is the major capsid protein of iridoviruses a suitable target for the study of viral evolution? Virus Genes 1998, 16, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 2001, 75, 11720–11734. [Google Scholar] [CrossRef]
- Larsen, J.B.; Larsen, A.; Bratbak, G.; Sandaa, R.A. Phylogenetic analysis of members of the phycodnaviridae virus family, using amplified fragments of the major capsid protein gene. Appl. Environ. Microb. 2008, 74, 3048–3057. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | SSR Viromes | |||
|---|---|---|---|---|
| D39s | DA4s | DA4b | ||
| Quality control | Raw reads | 4,2547,324 | 43,791,908 | 43,371,374 |
| Cut adaptor | 40,670,620 (95.59%) | 41,883,752 (95.64%) | 41,353,540 (95.35%) | |
| Q20 >20% | 35,357,306 (83.10%) | 37,567,224 (85.79%) | 38,397,082 (88.53%) | |
| Q30 >30% | 35,334,370 (83.05%) | 36,385,270 (83.09%) | 38,203,624 (88.08%) | |
| Assemble | All assembled contigs (>500 bp) | 2,418,081 | 3,699,559 | 1,693,019 |
| Non-redundancy contigs | 145,023 | 135,910 | 234,648 | |
| Mapping | Mapped reads | 15,307,526 (43.32%) | 12,760,519 (35.07%) | 21,197,289 (55.49%) |
| Group | Order | Family | Relative Abundance (%) | ||
|---|---|---|---|---|---|
| D39s | DA4s | DA4b | |||
| dsDNA | Caudovirales | Podoviridae | 41.92 | 48.70 | 42.15 |
| Caudovirales | Myoviridae | 28.34 | 22.92 | 29.46 | |
| Caudovirales | Siphoviridae | 11.92 | 13.38 | 14.08 | |
| Caudovirales | unclassified | 3.56 | 2.99 | 2.90 | |
| Caudovirales | Phycodnaviridae | 3.57 | 2.22 | 1.32 | |
| -- | Mimiviridae | 0.16 | 0.22 | 0.10 | |
| -- | Poxviridae | 0.14 | 0.22 | 1.13 | |
| -- | Iridoviridae | 0.37 | 0.44 | 0.30 | |
| unclassified | 3.13 | 2.84 | 3.17 | ||
| ssDNA | -- | Inoviridae | 0.10 | 0.13 | 0.12 |
| -- | Microviridae | 0.00 | 0.10 | 0.04 | |
| -- | Circoviridae | 0.01 | 0.11 | 0.01 | |
| -- | unclassified | 0.09 | 0.05 | 0.05 | |
| virophage | -- | 2.00 | 0.58 | 0.19 | |
| others | Ortervirales | Retroviridae | 0.26 | 0.29 | 0.21 |
| Ortervirales | Caulimoviridae | 0.07 | 0.76 | 0.04 | |
| Unclassified phage/viruses | -- | 3.71 | 3.01 | 4.07 | |
| Virome | MetaVir ID. | Total Sequenced Reads | Viral Hits (%) | Relative Abundance (%) | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dsDNA Viruses, No RNA Stage | RNA | ssDNA | Satellite | Virophage | |||||||||||||||||||||
| Ackermann | Baculo | Myo | Podo | Sipho | Herpes | Irido | Mimi | Papilloma | Phycodn | Polyom | Pox | Others | Circo | Gemini | Ino | Micro | Nano | Others | |||||||
| Antar Lake Spring | 10 | 41,322 | 20.51 | 0 | 0 | 0.05 | 0.06 | 0.22 | 0 | 0 | 0 | 0 | 0.01 | 0 | 0 | 0.03 | 0.14 | 22.00 | 0.27 | 0.03 | 3.45 | 0.22 | 23.39 | 6.82 | 0 |
| Antar Lake Summer | 11 | 38,475 | 22.07 | 0 | 0.01 | 0.32 | 0.30 | 0.72 | 0.03 | 0.01 | 0.38 | 0 | 1.57 | 0.02 | 0.09 | 0.20 | 0.40 | 61.96 | 0.49 | 0.11 | 1.31 | 0.93 | 15.52 | 3.20 | 0.19 |
| Lake Pavin | 6 | 649,290 | 25.04 | 0 | 0 | 1.60 | 1.29 | 6.73 | 0.01 | 0.01 | 0.01 | 0 | 0.06 | 0.01 | 0.02 | 0.85 | 28.04 | 6.02 | 0.07 | 0.12 | 1.79 | 0.06 | 17.07 | 0.93 | 0.25 |
| Lake Bourget | 7 | 593,084 | 37.46 | 0.01 | 0 | 2.30 | 2.98 | 11.56 | 0.02 | 0.01 | 0.01 | 0 | 0.14 | 0.02 | 0.06 | 1.86 | 0.28 | 1.77 | 0.03 | 0.01 | 60.35 | 0.32 | 1.99 | 0.36 | 0.20 |
| Tilapia Channel | 33 | 264,844 | 9.18 | 0.03 | 0.02 | 5.40 | 11.42 | 11.84 | 0.04 | 0.03 | 0.04 | 0 | 0.09 | 0.06 | 0 | 2.77 | 0.02 | 19.89 | 13.46 | 0.42 | 8.26 | 0.16 | 12.31 | 3.80 | 0.03 |
| Lough Neagh | 4925 | 2,295,055 | 24.65 | 0.14 | 0.08 | 10.96 | 32.59 | 36.71 | 0.11 | 0.08 | 0.02 | 0.06 | 0.38 | 0.05 | 0.31 | 16.11 | 0.32 | 0.01 | 0.03 | 0.16 | 0.04 | 0 | 0.39 | 0 | 0.13 |
| Jiulong River Estuary | 6305 | 498,957 | 31.40 | 0.05 | 0.06 | 14.67 | 32.36 | 34.80 | 0.12 | 0.04 | 0.11 | 0.02 | 0.79 | 0.16 | 0.49 | 13.58 | 0.27 | 0 | 0.01 | 0.11 | 0.01 | 0.01 | 0.22 | 0.31 | 0.14 |
| GS117 | 1479 | 480,375 | 38.80 | 0.04 | 0.03 | 16.50 | 35.59 | 27.05 | 0.06 | 0.04 | 0.06 | 0.01 | 0.53 | 0.06 | 0.44 | 17.59 | 0.17 | 0.01 | 0.01 | 0.02 | 0.01 | 0.01 | 0.07 | 0.32 | 0.28 |
| M1CS | 1440 | 303,519 | 32.93 | 0.05 | 0.03 | 10.92 | 28.08 | 48.75 | 0.09 | 0.06 | 0.06 | 0.02 | 0.47 | 0.07 | 0.15 | 9.85 | 0.11 | 0.03 | 0.01 | 0.01 | 0 | 0.04 | 0.11 | 0.29 | 0.11 |
| Dunk Island | 1357 | 1,165,256 | 3.61 | 0.01 | 0.02 | 12.61 | 40.74 | 21.80 | 0.19 | 0.04 | 0.04 | 0.06 | 0.36 | 0.04 | 0.15 | 22.43 | 0.09 | 0 | 0.05 | 0.02 | 0 | 0 | 0.07 | 0.53 | 0.20 |
| Fitzroy Island | 1358 | 82,739 | 32.66 | 0.02 | 0.05 | 13.33 | 48.00 | 17.30 | 0.04 | 0.05 | 0.05 | 0.06 | 0.44 | 0.12 | 0.01 | 19.36 | 0.20 | 0 | 0.04 | 0.04 | 0 | 0 | 0.09 | 0 | 0.29 |
| LA26S | 1396 | 165,256 | 28.45 | 0.04 | 0.17 | 15.51 | 44.05 | 24.32 | 0.08 | 0.10 | 0.11 | 0.02 | 1.17 | 0.27 | 0.27 | 11.76 | 0.10 | 0 | 0.02 | 0.07 | 0 | 0 | 0.21 | 0.76 | 0.19 |
| D39s | -- | 17,667,185 | 11.97 | 0.09 | 0.06 | 28.33 | 41.90 | 11.92 | 0.03 | 0.33 | 0.16 | 0 | 3.57 | 0.05 | 0.14 | 7.04 | 0.04 | 0.01 | 0 | 0.07 | 0 | 0 | 0.10 | 0 | 2.00 |
| DA4s | -- | 19,101,812 | 5.09 | 0.06 | 0.22 | 22.91 | 48.68 | 13.37 | 0.05 | 0.41 | 0.22 | 0 | 2.21 | 0.06 | 0.22 | 6.33 | 0.10 | 0.11 | 0 | 0.10 | 0.10 | 0 | 0.07 | 0 | 0.58 |
| DA4b | -- | 18,192,635 | 13.87 | 0.10 | 0.05 | 29.46 | 42.14 | 14.08 | 0.02 | 0.26 | 0.10 | 0 | 1.32 | 0.01 | 1.13 | 6.44 | 0.05 | 0.01 | 0 | 0.09 | 0.04 | 0 | 0.07 | 0 | 0.19 |
| Sargasso Sea | 12 | 397,939 | 11.80 | 0.01 | 0 | 2.20 | 6.32 | 2.48 | 0 | 0.02 | 0.02 | 0 | 0.08 | 0.04 | 0 | 2.88 | 0.01 | 14.21 | 0 | 0 | 43.69 | 0.03 | 24.83 | 0.60 | 0.01 |
| Arctic Vir | 15 | 686,209 | 1.30 | 0.04 | 0.04 | 9.91 | 9.63 | 23.14 | 0.54 | 0.08 | 0.25 | 0.05 | 1.24 | 1.37 | 0.76 | 3.19 | 0.99 | 0.64 | 0.04 | 5.18 | 0 | 0 | 1.88 | 16.07 | 0.06 |
| Arctic Ocean | 1158 | 79,646 | 14.15 | 0 | 0 | 0.12 | 0.12 | 0.39 | 0 | 0 | 0.01 | 0 | 0.03 | 0.01 | 0.02 | 0.09 | 0.32 | 63.14 | 0.09 | 0.16 | 2.17 | 0.19 | 13.93 | 4.25 | 0 |
| Black Sea | 1155 | 78436 | 16.47 | 0 | 0 | 0.05 | 0.03 | 0.08 | 0 | 0 | 0 | 0 | 0.02 | 0 | 0.01 | 0.02 | 0.12 | 56.33 | 0.60 | 0.01 | 0.88 | 0.17 | 14.24 | 5.79 | 0 |
| Med Sea | 1161 | 65,340 | 16.13 | 0 | 0.01 | 2.29 | 0.82 | 2.49 | 0.04 | 0.05 | 0.02 | 0.01 | 0.16 | 0.04 | 1.58 | 0.35 | 0.29 | 21.05 | 0 | 0 | 59.09 | 0 | 2.96 | 7.51 | 0 |
| OMZst3viral10m | 897 | 128,441 | 15.56 | 0.01 | 0.01 | 2.90 | 4.00 | 2.12 | 0.01 | 0.02 | 0.04 | 0 | 0.40 | 0.03 | 0.06 | 1.07 | 0.08 | 60.79 | 0 | 0.03 | 6.49 | 0.13 | 14.89 | 1.88 | 0.16 |
| Antar hypolith | 2726 | 1,057,535 | 12.74 | 0.01 | 0.01 | 1.31 | 1.47 | 7.15 | 0.13 | 0.01 | 0 | 0.05 | 0.02 | 0.03 | 0.02 | 0.36 | 0.13 | 0.03 | 0 | 0.21 | 88.42 | 0 | 0.25 | 0.09 | 0 |
| Antar open soil | 2727 | 870,687 | 16.99 | 0.08 | 0.05 | 9.25 | 4.96 | 16.74 | 0.29 | 0.05 | 0.03 | 0.06 | 0.24 | 0.05 | 0.09 | 3.10 | 0.43 | 0.01 | 0 | 0.03 | 63.28 | 0 | 0.40 | 0.07 | 0 |
| Biome | Virome | MetaVir Project ID. | Number of Reads | South Scotia Sea | ||
|---|---|---|---|---|---|---|
| D39s | DA4s | DA4b | ||||
| Antarctic seawater | D39s | - | 35,334,370 | 100% | 50.35% | 49.60% |
| Antarctic seawater | DA4s | - | 36,385,270 | 48.90% | 100% | 28.37% |
| Antarctic seawater | DA4b | - | 38,203,624 | 45.88% | 27.02% | 100% |
| Seawater | OMZst3viral10m | 897 | 128,441 | 6.50% | 6.86% | 10.69% |
| Seawater | GS117 | 1479 | 480,375 | 8.61% | 9.09% | 14.57% |
| Arctic seawater | Arctic Vir | 15 | 686,209 | 1.45% | 1.33% | 2.10% |
| Seawater | Sargasso Sea | 12 | 397,939 | 4.68% | 4.97% | 7.69% |
| POV seawater | Dunk Island | 1357 | 1,165,256 | 0.55% | 0.71% | 1.14% |
| POV seawater | Fitzroy Island | 1358 | 82,739 | 7.44% | 9.11% | 14.04% |
| POV seawater | LA26S | 1396 | 165,256 | 16.75% | 14.72% | 21.02% |
| POV seawater | M1CS | 1440 | 303,519 | 14.77% | 15.59% | 23.12% |
| Deep Ocean | Arctic Ocean | 1158 | 79,646 | 1.64% | 2.17% | 4.22% |
| Deep Ocean | Black Sea | 1155 | 78,436 | 0.57% | 0.62% | 0.69% |
| Deep Ocean | Med Sea | 1161 | 65,340 | 0.86% | 0.89% | 2.04% |
| Freshwater | Lake Bourget | 7 | 593,084 | 0.94% | 0.87% | 1.66% |
| Freshwater | Lake Pavin | 6 | 649,290 | 0.25% | 0.23% | 0.44% |
| Antarctic freshwater | Antar Lake Spring | 10 | 41,322 | 0.07% | 0.06% | 0.16% |
| Antarctic freshwater | Antar Lake Summer | 11 | 38,475 | 0.41% | 0.43% | 1.87% |
| Freshwater | Lough Neagh | 4925 | 2,295,055 | 0.31% | 0.30% | 0.52% |
| Freshwater | Jiulong River Estuary | 6305 | 498,957 | 5.74% | 6.06% | 9.61% |
| Freshwater | Tilapia Channel | 33 | 264,844 | 0.14% | 0.15% | 0.31% |
| Antarctic soil | Antar open soil | 2727 | 870,687 | 0.43% | 0.42% | 0.59% |
| Antarctic hypolith | Antar hypolith | 2726 | 1,057,535 | 0.26% | 0.42% | 0.36% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Gao, C.; Jiang, Y.; Wang, M.; Zhou, X.; Shao, H.; Gong, Z.; McMinn, A. Metagenomic Characterization of the Viral Community of the South Scotia Ridge. Viruses 2019, 11, 95. https://doi.org/10.3390/v11020095
Yang Q, Gao C, Jiang Y, Wang M, Zhou X, Shao H, Gong Z, McMinn A. Metagenomic Characterization of the Viral Community of the South Scotia Ridge. Viruses. 2019; 11(2):95. https://doi.org/10.3390/v11020095
Chicago/Turabian StyleYang, Qingwei, Chen Gao, Yong Jiang, Min Wang, Xinhao Zhou, Hongbing Shao, Zheng Gong, and Andrew McMinn. 2019. "Metagenomic Characterization of the Viral Community of the South Scotia Ridge" Viruses 11, no. 2: 95. https://doi.org/10.3390/v11020095
APA StyleYang, Q., Gao, C., Jiang, Y., Wang, M., Zhou, X., Shao, H., Gong, Z., & McMinn, A. (2019). Metagenomic Characterization of the Viral Community of the South Scotia Ridge. Viruses, 11(2), 95. https://doi.org/10.3390/v11020095