In vitro Modeling of Prion Strain Tropism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
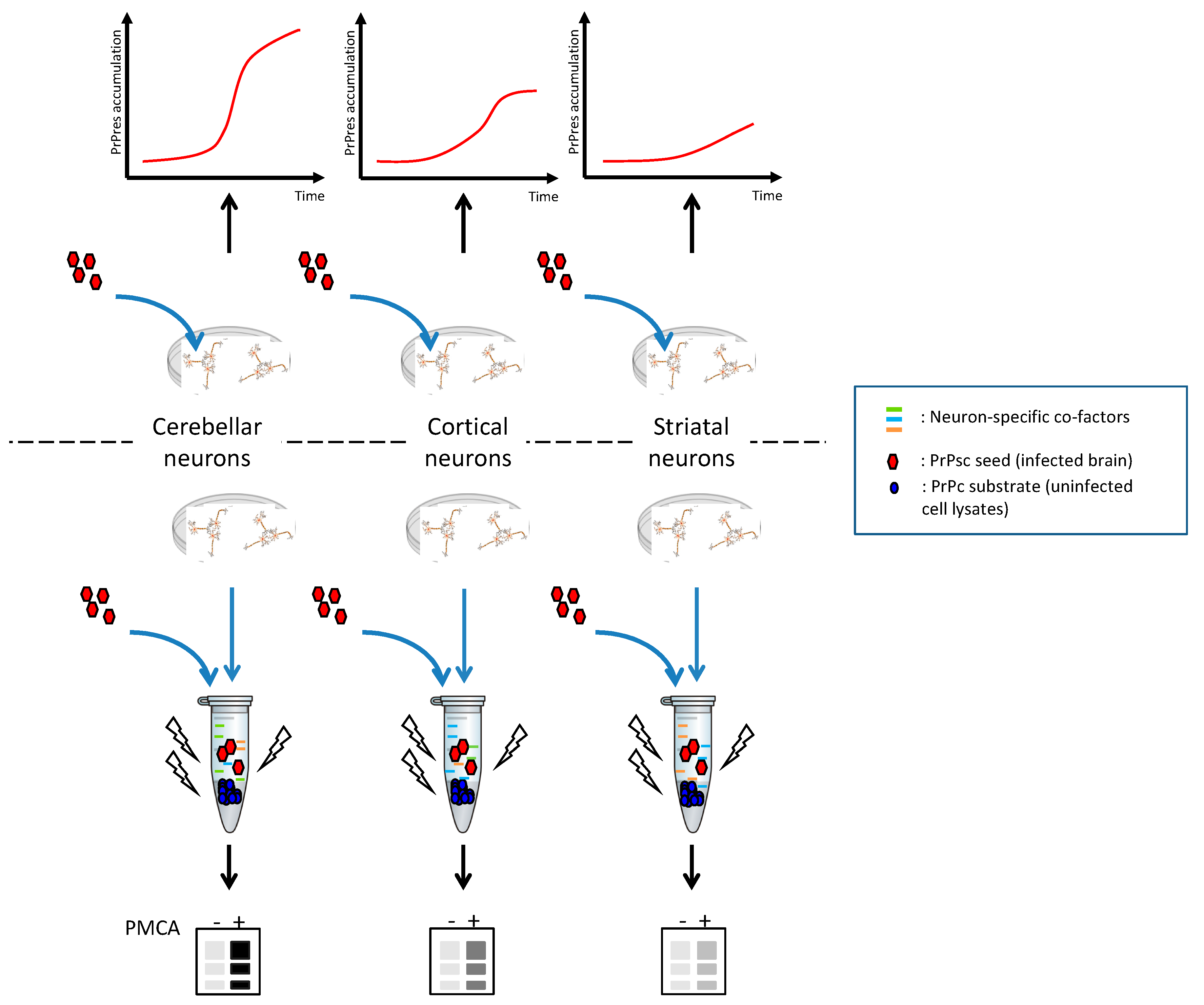
2. Cellular Models of Prion Strain Tropism
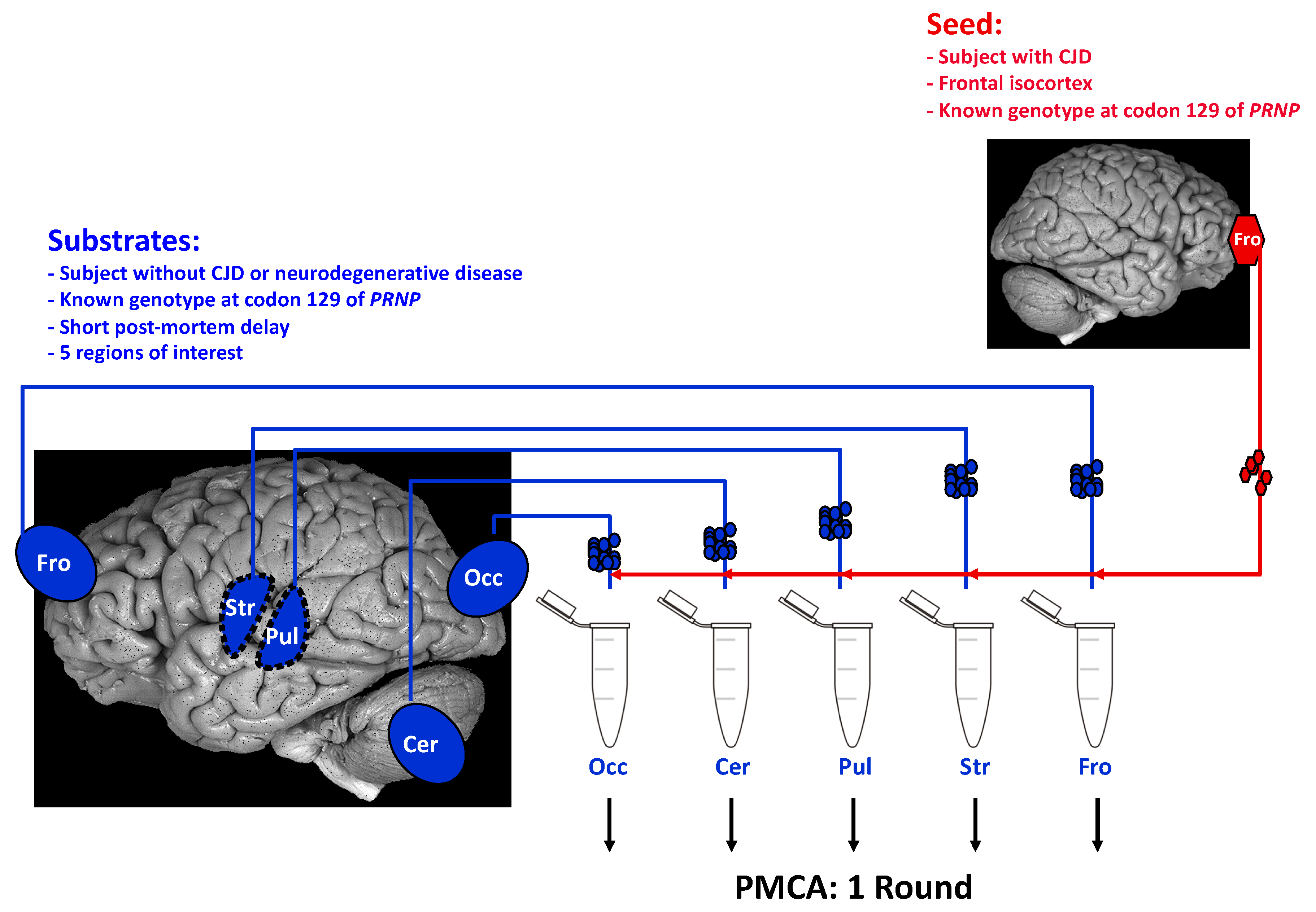
3. Acellular Models of Prion Strain Tropism
4. Combining Cellular with Acellular Models
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Zou, W.; Wang, W.; Brown, P.; Capellari, S.; Ghetti, B.; Kopp, N.; Schulz-Schaeffer, W.J.; Kretzschmar, H.A.; Head, M.W.; et al. Genetic influence on the structural variations of the abnormal prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 10168–10172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parchi, P.; Castellani, R.; Capellari, S.; Ghetti, B.; Young, K.; Chen, S.G.; Farlow, M.; Dickson, D.W.; Sima, A.A.; Trojanowski, J.Q.; et al. Molecular basis of phenotypic variability in sporadic creutzfeldt-jakob disease. Ann. Neurol. 1996, 39, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic creutzfeldt-jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Parchi, P.; Notari, S.; Weber, P.; Schimmel, H.; Budka, H.; Ferrer, I.; Haik, S.; Hauw, J.J.; Head, M.W.; Ironside, J.W.; et al. Inter-laboratory assessment of prpsc typing in creutzfeldt-jakob disease: A western blot study within the neuroprion consortium. Brain Pathol. 2009, 19, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Joiner, S.; Wadsworth, J.D.; Sidle, K.C.; Bell, J.E.; Budka, H.; Ironside, J.W.; Collinge, J. Molecular classification of sporadic creutzfeldt-jakob disease. Brain 2003, 126, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Rudge, P.; Jaunmuktane, Z.; Adlard, P.; Bjurstrom, N.; Caine, D.; Lowe, J.; Norsworthy, P.; Hummerich, H.; Druyeh, R.; Wadsworth, J.D.; et al. Iatrogenic cjd due to pituitary-derived growth hormone with genetically determined incubation times of up to 40 years. Brain 2015, 138, 3386–3399. [Google Scholar] [CrossRef]
- Parchi, P.; de Boni, L.; Saverioni, D.; Cohen, M.L.; Ferrer, I.; Gambetti, P.; Gelpi, E.; Giaccone, G.; Hauw, J.J.; Hoftberger, R.; et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: An inter-rater study among surveillance centres in europe and USA. Acta Neuropathol. 2012, 124, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A. Staining, straining and restraining prions. Nat. Neurosci. 2008, 11, 1239–1240. [Google Scholar] [CrossRef]
- Pattison, I.H.; Millson, G.C. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. Ther. 1961, 71, 101–108. [Google Scholar] [CrossRef]
- Bruce, M.E. Scrapie strain variation and mutation. Br. Med. Bull. 1993, 49, 822–838. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that ‘new variant’ cjd is caused by the bse agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Casalone, C.; Zanusso, G.; Acutis, P.; Ferrari, S.; Capucci, L.; Tagliavini, F.; Monaco, S.; Caramelli, M. Identification of a second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic creutzfeldt-jakob disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3065–3070. [Google Scholar] [CrossRef] [PubMed]
- Biacabe, A.G.; Laplanche, J.L.; Ryder, S.; Baron, T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep. 2004, 5, 110–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, E.S.; Young, S. Chronic wasting disease of captive mule deer: A spongiform encephalopathy. J. Wildl. Dis. 1980, 16, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Telling, G.C. Chronic wasting disease: An evolving prion disease of cervids. Handb. Clin. Neurol. 2018, 153, 135–151. [Google Scholar] [PubMed]
- Bishop, M.T.; Will, R.G.; Manson, J.C. Defining sporadic creutzfeldt-jakob disease strains and their transmission properties. Proc. Natl. Acad. Sci. USA 2010, 107, 12005–12010. [Google Scholar] [CrossRef]
- Diack, A.B.; Ritchie, D.; Bishop, M.; Pinion, V.; Brandel, J.P.; Haik, S.; Tagliavini, F.; Van Duijn, C.; Belay, E.D.; Gambetti, P.; et al. Constant transmission properties of variant creutzfeldt-jakob disease in 5 countries. Emerg. Infect. Dis. 2012, 18, 1574–1579. [Google Scholar] [CrossRef]
- Haik, S.; Brandel, J.P. Biochemical and strain properties of cjd prions: Complexity versus simplicity. J. Neurochem. 2011, 119, 251–261. [Google Scholar] [CrossRef]
- Bruce, M.E. Serial studies on the development of cerebral amyloidosis and vacuolar degeneration in murine scrapie. J. Comp. Pathol. Ther. 1981, 91, 589–597. [Google Scholar] [CrossRef]
- Kim, Y.S.; Carp, R.L.; Callahan, S.M.; Wisniewski, H.M. Incubation periods and survival times for mice injected stereotaxically with three scrapie strains in different brain regions. J. Gen. Virol. 1987, 68, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Cescatti, M.; Notari, S.; Schulz-Schaeffer, W.J.; Capellari, S.; Giese, A.; Zou, W.Q.; Kretzschmar, H.; Ghetti, B.; Brown, P. Agent strain variation in human prion disease: Insights from a molecular and pathological review of the national institutes of health series of experimentally transmitted disease. Brain 2010, 133, 3030–3042. [Google Scholar] [CrossRef] [PubMed]
- Haik, S.; Brandel, J.P. Infectious prion diseases in humans: Cannibalism, iatrogenicity and zoonoses. Infect. Genet. Evol. 2014, 26, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Brandel, J.P.; Heath, C.A.; Head, M.W.; Levavasseur, E.; Knight, R.; Laplanche, J.L.; Langeveld, J.P.; Ironside, J.W.; Hauw, J.J.; Mackenzie, J.; et al. Variant creutzfeldt-jakob disease in france and the united kingdom: Evidence for the same agent strain. Ann. Neurol. 2009, 65, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Kincaid, A.E.; Bartz, J.C. Prion strain targeting independent of strain-specific neuronal tropism. J. Virol. 2009, 83, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Ryou, C.; Mays, C.E. Prion propagation in vitro: Are we there yet? Int. J. Med. Sci. 2008, 5, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Klohn, P.C.; Stoltze, L.; Flechsig, E.; Enari, M.; Weissmann, C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad. Sci. USA 2003, 100, 11666–11671. [Google Scholar] [CrossRef] [Green Version]
- Cronier, S.; Laude, H.; Peyrin, J.M. Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death. Proc. Natl. Acad. Sci. USA 2004, 101, 12271–12276. [Google Scholar] [CrossRef] [Green Version]
- Cronier, S.; Beringue, V.; Bellon, A.; Peyrin, J.M.; Laude, H. Prion strain- and species-dependent effects of antiprion molecules in primary neuronal cultures. J. Virol. 2007, 81, 13794–13800. [Google Scholar] [CrossRef]
- Falsig, J.; Sonati, T.; Herrmann, U.S.; Saban, D.; Li, B.; Arroyo, K.; Ballmer, B.; Liberski, P.P.; Aguzzi, A. Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS Pathog. 2012, 8, e1002985. [Google Scholar] [CrossRef]
- Hannaoui, S.; Maatouk, L.; Privat, N.; Levavasseur, E.; Faucheux, B.A.; Haik, S. Prion propagation and toxicity occur in vitro with two-phase kinetics specific to strain and neuronal type. J. Virol. 2013, 87, 2535–2548. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; Clarke, A.R.; Collinge, J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011, 470, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Hannaoui, S.; Gougerot, A.; Privat, N.; Levavasseur, E.; Bizat, N.; Hauw, J.J.; Brandel, J.P.; Haik, S. Cycline efficacy on the propagation of human prions in primary cultured neurons is strain-specific. J. Infect. Dis. 2014, 209, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Krejciova, Z.; Alibhai, J.; Zhao, C.; Krencik, R.; Rzechorzek, N.M.; Ullian, E.M.; Manson, J.; Ironside, J.W.; Head, M.W.; Chandran, S. Human stem cell-derived astrocytes replicate human prions in a prnp genotype-dependent manner. J. Exp. Med. 2017, 214, 3481–3495. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.; Barres, B. Snapshot: Astrocytes in health and disease. Cell 2015, 162, 1170. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocisko, D.A.; Come, J.H.; Priola, S.A.; Chesebro, B.; Raymond, G.J.; Lansbury, P.T.; Caughey, B. Cell-free formation of protease-resistant prion protein. Nature 1994, 370, 471–474. [Google Scholar] [CrossRef] [Green Version]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef]
- Castilla, J.; Saa, P.; Hetz, C.; Soto, C. In vitro generation of infectious scrapie prions. Cell 2005, 121, 195–206. [Google Scholar] [CrossRef]
- Castilla, J.; Saa, P.; Morales, R.; Abid, K.; Maundrell, K.; Soto, C. Protein misfolding cyclic amplification for diagnosis and prion propagation studies. Methods Enzymol. 2006, 412, 3–21. [Google Scholar]
- Klingeborn, M.; Race, B.; Meade-White, K.D.; Chesebro, B. Lower specific infectivity of protease-resistant prion protein generated in cell-free reactions. Proc. Natl. Acad. Sci. USA 2011, 108, E1244–E1253. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.; Giese, A.; Piening, N.; Mitteregger, G.; Thomzig, A.; Beekes, M.; Kretzschmar, H.A. Cell-free formation of misfolded prion protein with authentic prion infectivity. Proc. Natl. Acad. Sci. USA 2006, 103, 15818–15823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saa, P.; Castilla, J.; Soto, C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 2006, 281, 35245–35252. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Peden, A.H.; Prowse, C.V.; Groner, A.; Manson, J.C.; Turner, M.L.; Ironside, J.W.; MacGregor, I.R.; Head, M.W. In vitro amplification and detection of variant creutzfeldt-jakob disease prpsc. J. Pathol. 2007, 213, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Peden, A.H.; Wight, D.; Prowse, C.; Macgregor, I.; Manson, J.; Turner, M.; Ironside, J.W.; Head, M.W. Effects of human prpsc type and prnp genotype in an in-vitro conversion assay. Neuroreport 2008, 19, 1783–1786. [Google Scholar] [CrossRef]
- Privat, N.; Levavasseur, E.; Yildirim, S.; Hannaoui, S.; Brandel, J.P.; Laplanche, J.L.; Beringue, V.; Seilhean, D.; Haik, S. Region-specific protein misfolding cyclic amplification reproduces brain tropism of prion strains. J. Biol. Chem. 2017, 292, 16688–16696. [Google Scholar] [CrossRef]
- Castilla, J.; Gonzalez-Romero, D.; Saa, P.; Morales, R.; De Castro, J.; Soto, C. Crossing the species barrier by prp(sc) replication in vitro generates unique infectious prions. Cell 2008, 134, 757–768. [Google Scholar] [CrossRef]
- Green, K.M.; Castilla, J.; Seward, T.S.; Napier, D.L.; Jewell, J.E.; Soto, C.; Telling, G.C. Accelerated high fidelity prion amplification within and across prion species barriers. PLoS Pathog. 2008, 4, e1000139. [Google Scholar] [CrossRef]
- Fernandez-Borges, N.; de Castro, J.; Castilla, J. In vitro studies of the transmission barrier. Prion 2009, 3, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.; Wight, D.; Barron, R.; Jeffrey, M.; Manson, J.; Prowse, C.; Ironside, J.W.; Head, M.W. Molecular model of prion transmission to humans. Emerg. Infect. Dis. 2009, 15, 2013–2016. [Google Scholar] [CrossRef]
- Jones, M.; Peden, A.H.; Head, M.W.; Ironside, J.W. The application of in vitro cell-free conversion systems to human prion diseases. Acta Neuropathol. 2010, 121, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.E.; Thorne, L.; Lockey, R.; Vickery, C.M.; Terry, L.A.; Bujdoso, R.; Spiropoulos, J. Strain typing of classical scrapie by transgenic mouse bioassay using protein misfolding cyclic amplification to replace primary passage. PLoS ONE 2013, 8, e57851. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Saa, P.; Soto, C. Detection of prions in blood. Nat. Med. 2005, 11, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Saa, P.; Castilla, J.; Soto, C. Presymptomatic detection of prions in blood. Science 2006, 313, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Yoshioka, M.; Okada, H.; Takata, M.; Yokoyama, T.; Mohri, S. Urinary excretion and blood level of prions in scrapie-infected hamsters. J. Gen. Virol. 2007, 88, 2890–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Romero, D.; Barria, M.A.; Leon, P.; Morales, R.; Soto, C. Detection of infectious prions in urine. FEBS Lett. 2008, 582, 3161–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, N.J.; Seelig, D.M.; Zabel, M.D.; Telling, G.C.; Hoover, E.A. Detection of cwd prions in urine and saliva of deer by transgenic mouse bioassay. PLoS ONE 2009, 4, e4848. [Google Scholar] [CrossRef]
- Tattum, M.H.; Jones, S.; Pal, S.; Collinge, J.; Jackson, G.S. Discrimination between prion-infected and normal blood samples by protein misfolding cyclic amplification. Transfusion 2010, 50, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [Green Version]
- Barria, M.A.; Mukherjee, A.; Gonzalez-Romero, D.; Morales, R.; Soto, C. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog. 2009, 5, e1000421. [Google Scholar] [CrossRef]
- Wang, F.; Wang, X.; Yuan, C.G.; Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. Rna molecules stimulate prion protein conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef]
- Nishina, K.A.; Deleault, N.R.; Mahal, S.P.; Baskakov, I.; Luhrs, T.; Riek, R.; Supattapone, S. The stoichiometry of host prpc glycoforms modulates the efficiency of prpsc formation in vitro. Biochemistry 2006, 45, 14129–14139. [Google Scholar] [CrossRef] [PubMed]
- Orem, N.R.; Geoghegan, J.C.; Deleault, N.R.; Kascsak, R.; Supattapone, S. Copper (ii) ions potently inhibit purified prpres amplification. J. Neurochem. 2006, 96, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.C.; Valdes, P.A.; Orem, N.R.; Deleault, N.R.; Williamson, R.A.; Harris, B.T.; Supattapone, S. Selective incorporation of polyanionic molecules into hamster prions. J. Biol. Chem. 2007, 282, 36341–36353. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Peden, A.H.; Yull, H.; Wight, D.; Bishop, M.T.; Prowse, C.V.; Turner, M.L.; Ironside, J.W.; MacGregor, I.R.; Head, M.W. Human platelets as a substrate source for the in vitro amplification of the abnormal prion protein (prp) associated with variant creutzfeldt-jakob disease. Transfusion 2009, 49, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Titlow, W.; Seward, T.; Telling, G.C.; Ryou, C. Enhancement of protein misfolding cyclic amplification by using concentrated cellular prion protein source. Biochem. Biophys. Res. Commun. 2009, 388, 306–310. [Google Scholar] [CrossRef] [Green Version]
- Abid, K.; Morales, R.; Soto, C. Cellular factors implicated in prion replication. FEBS Lett. 2010, 584, 2409–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deleault, N.R.; Kascsak, R.; Geoghegan, J.C.; Supattapone, S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 2010, 49, 3928–3934. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Montalban, N.; Makarava, N.; Ostapchenko, V.G.; Savtchenk, R.; Alexeeva, I.; Rohwer, R.G.; Baskakov, I.V. Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 2011, 7, e1001277. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Ryou, C. Plasminogen stimulates propagation of protease-resistant prion protein in vitro. FASEB J. 2010, 24, 5102–5112. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Ryou, C. Plasminogen: A cellular protein cofactor for prpsc propagation. Prion 2011, 5, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Harris, B.T.; Supattapone, S. In situ photodegradation of incorporated polyanion does not alter prion infectivity. PLoS Pathog. 2011, 7, e1002001. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.P.; Morales, R.; Duran-Aniotz, C.; Moreno-Gonzalez, I.; Khan, U.; Soto, C. Role of prion replication in the strain-dependent brain regional distribution of prions. J. Biol. Chem. 2016, 291, 12880–12887. [Google Scholar] [CrossRef] [PubMed]
- Klemm, H.M.; Welton, J.M.; Masters, C.L.; Klug, G.M.; Boyd, A.; Hill, A.F.; Collins, S.J.; Lawson, V.A. The prion protein preference of sporadic creutzfeldt-jakob disease subtypes. J. Biol. Chem. 2012, 287, 36465–36472. [Google Scholar] [CrossRef] [PubMed]
- Lawson, V.A.; Lumicisi, B.; Welton, J.; Machalek, D.; Gouramanis, K.; Klemm, H.M.; Stewart, J.D.; Masters, C.L.; Hoke, D.E.; Collins, S.J.; et al. Glycosaminoglycan sulphation affects the seeded misfolding of a mutant prion protein. PLoS ONE 2010, 5, e12351. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Flechsig, E.; Weissmann, C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. USA 2001, 98, 9295–9299. [Google Scholar] [CrossRef]
- Priola, S.A.; Lawson, V.A. Glycosylation influences cross-species formation of protease-resistant prion protein. EMBO J. 2001, 20, 6692–6699. [Google Scholar] [CrossRef] [Green Version]
- Levavasseur, E.; Laffont-Proust, I.; Morain, E.; Faucheux, B.A.; Privat, N.; Peoc’h, K.; Sazdovitch, V.; Brandel, J.P.; Hauw, J.J.; Haik, S. Regulating factors of prp glycosylation in creutzfeldt-jakob disease—Implications for the dissemination and the diagnosis of human prion strains. PLoS ONE 2008, 3, e2786. [Google Scholar] [CrossRef]
- Cancellotti, E.; Bradford, B.M.; Tuzi, N.L.; Hickey, R.D.; Brown, D.; Brown, K.L.; Barron, R.M.; Kisielewski, D.; Piccardo, P.; Manson, J.C. Glycosylation of prpc determines timing of neuroinvasion and targeting in the brain following transmissible spongiform encephalopathy infection by a peripheral route. J. Virol. 2010, 84, 3464–3475. [Google Scholar] [CrossRef]
- Tuzi, N.L.; Cancellotti, E.; Baybutt, H.; Blackford, L.; Bradford, B.; Plinston, C.; Coghill, A.; Hart, P.; Piccardo, P.; Barron, R.M.; et al. Host prp glycosylation: A major factor determining the outcome of prion infection. PLoS Biol. 2008, 6, e100. [Google Scholar] [CrossRef] [PubMed]
- Saa, P.; Sferrazza, G.F.; Ottenberg, G.; Oelschlegel, A.M.; Dorsey, K.; Lasmezas, C.I. Strain-specific role of rnas in prion replication. J. Virol. 2012, 86, 10494–10504. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Gonzalez-Montalban, N.; Makarava, N.; Kovacs, G.G.; Baskakov, I.V. Prion replication environment defines the fate of prion strain adaptation. PLoS Pathog. 2018, 14, e1007093. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.D.; Wight, T.N.; Nochlin, D.; Koike, Y.; Kimata, K.; DeArmond, S.J.; Prusiner, S.B. Immunolocalization of heparan sulfate proteoglycans to the prion protein amyloid plaques of gerstmann-straussler syndrome, creutzfeldt-jakob disease and scrapie. Lab. Investig. 1990, 63, 601–611. [Google Scholar] [PubMed]
- Rieger, R.; Edenhofer, F.; Lasmezas, C.I.; Weiss, S. The human 37-kda laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat. Med. 1997, 3, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Gauczynski, S.; Peyrin, J.M.; Haik, S.; Leucht, C.; Hundt, C.; Rieger, R.; Krasemann, S.; Deslys, J.P.; Dormont, D.; Lasmezas, C.I.; et al. The 37-kda/67-kda laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001, 20, 5863–5875. [Google Scholar] [CrossRef] [PubMed]
- Hundt, C.; Peyrin, J.M.; Haik, S.; Gauczynski, S.; Leucht, C.; Rieger, R.; Riley, M.L.; Deslys, J.P.; Dormont, D.; Lasmezas, C.I.; et al. Identification of interaction domains of the prion protein with its 37-kda/67-kda laminin receptor. EMBO J. 2001, 20, 5876–5886. [Google Scholar] [CrossRef]
- Fernandez-Borges, N.; Di Bari, M.A.; Erana, H.; Sanchez-Martin, M.; Pirisinu, L.; Parra, B.; Elezgarai, S.R.; Vanni, I.; Lopez-Moreno, R.; Vaccari, G.; et al. Cofactors influence the biological properties of infectious recombinant prions. Acta Neuropathol. 2018, 135, 179–199. [Google Scholar] [CrossRef]
- Makarava, N.; Savtchenko, R.; Lasch, P.; Beekes, M.; Baskakov, I.V. Preserving prion strain identity upon replication of prions in vitro using recombinant prion protein. Acta Neuropathol. Commun. 2018, 6, 92. [Google Scholar] [CrossRef]
- Wadsworth, J.D.; Hill, A.F.; Joiner, S.; Jackson, G.S.; Clarke, A.R.; Collinge, J. Strain-specific prion-protein conformation determined by metal ions. Nat. Cell Biol. 1999, 1, 55–59. [Google Scholar] [CrossRef]
- Nishina, K.; Deleault, N.R.; Lucassen, R.W.; Supattapone, S. In vitro prion protein conversion in detergent-solubilized membranes. Biochemistry 2004, 43, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.C.; Miller, M.B.; Kwak, A.H.; Harris, B.T.; Supattapone, S. Trans-dominant inhibition of prion propagation in vitro is not mediated by an accessory cofactor. PLoS Pathog. 2009, 5, e1000535. [Google Scholar] [CrossRef]
- Kim, J.I.; Cali, I.; Surewicz, K.; Kong, Q.; Raymond, G.J.; Atarashi, R.; Race, B.; Qing, L.; Gambetti, P.; Caughey, B.; et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 2010, 285, 14083–14087. [Google Scholar] [CrossRef]
- Mays, C.E.; Yeom, J.; Kang, H.E.; Bian, J.; Khaychuk, V.; Kim, Y.; Bartz, J.C.; Telling, G.C.; Ryou, C. In vitro amplification of misfolded prion protein using lysate of cultured cells. PLoS ONE 2011, 6, e18047. [Google Scholar] [CrossRef] [PubMed]
- Saborio, G.P.; Soto, C.; Kascsak, R.J.; Levy, E.; Kascsak, R.; Harris, D.A.; Frangione, B. Cell-lysate conversion of prion protein into its protease-resistant isoform suggests the participation of a cellular chaperone. Biochem. Biophys. Res. Commun. 1999, 258, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Takeuchi, A.; Yamamoto, M.; Kitamoto, T.; Ironside, J.W.; Morita, M. Heparin enhances the cell-protein misfolding cyclic amplification efficiency of variant creutzfeldt-jakob disease. Neurosci. Lett. 2011, 498, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.A.; Lu, Z.Y.; Zaitsev, I.; Manuelidis, L. Microglial activation varies in different models of creutzfeldt-jakob disease. J. Virol. 1999, 73, 5089–5097. [Google Scholar]
- Falsig, J.; Julius, C.; Margalith, I.; Schwarz, P.; Heppner, F.L.; Aguzzi, A. A versatile prion replication assay in organotypic brain slices. Nat. Neurosci. 2008, 11, 109–117. [Google Scholar] [CrossRef]
- Nieznanski, K. Interactions of prion protein with intracellular proteins: So many partners and no consequences? Cell. Mol. Neurobiol. 2010, 30, 653–666. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levavasseur, E.; Privat, N.; Haïk, S. In vitro Modeling of Prion Strain Tropism. Viruses 2019, 11, 236. https://doi.org/10.3390/v11030236
Levavasseur E, Privat N, Haïk S. In vitro Modeling of Prion Strain Tropism. Viruses. 2019; 11(3):236. https://doi.org/10.3390/v11030236
Chicago/Turabian StyleLevavasseur, Etienne, Nicolas Privat, and Stéphane Haïk. 2019. "In vitro Modeling of Prion Strain Tropism" Viruses 11, no. 3: 236. https://doi.org/10.3390/v11030236
APA StyleLevavasseur, E., Privat, N., & Haïk, S. (2019). In vitro Modeling of Prion Strain Tropism. Viruses, 11(3), 236. https://doi.org/10.3390/v11030236