Molecular Characterization and Evolutionary Analyses of Carnivore Protoparvovirus 1 NS1 Gene

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Parvovirus PCR
2.3. Viral Isolation
2.4. Sequence Analysis
2.5. Recombination and Selection Pressure Analyses
2.6. Phylogenetic Analysis
3. Results
3.1. Detection and Characterization of FPLV and CPV
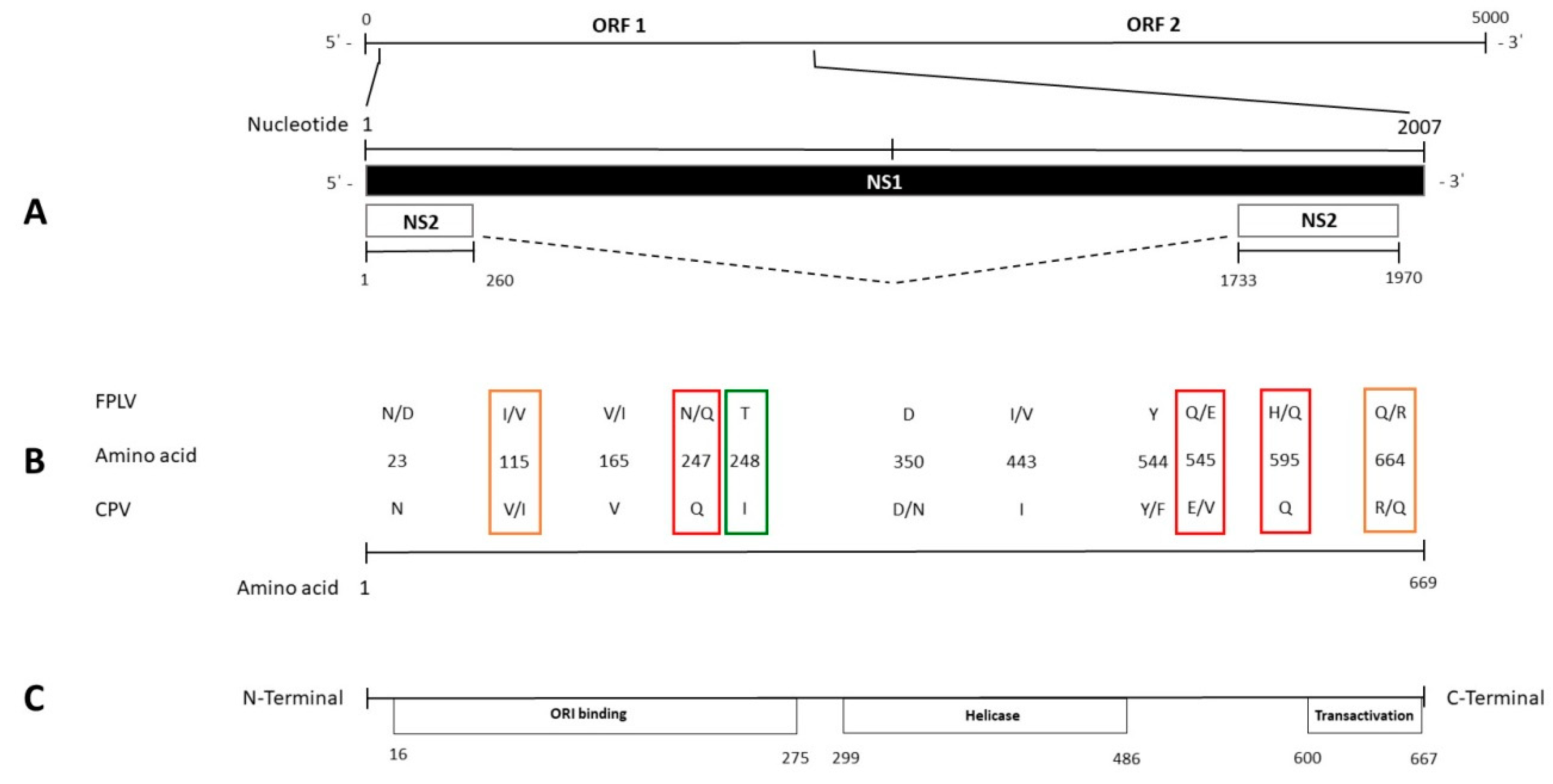
3.2. Sequence Analysis of NS1 Gene
3.3. Sequence Analysis of NS2 Gene
3.4. Recombination and Selection Pressure Analyses
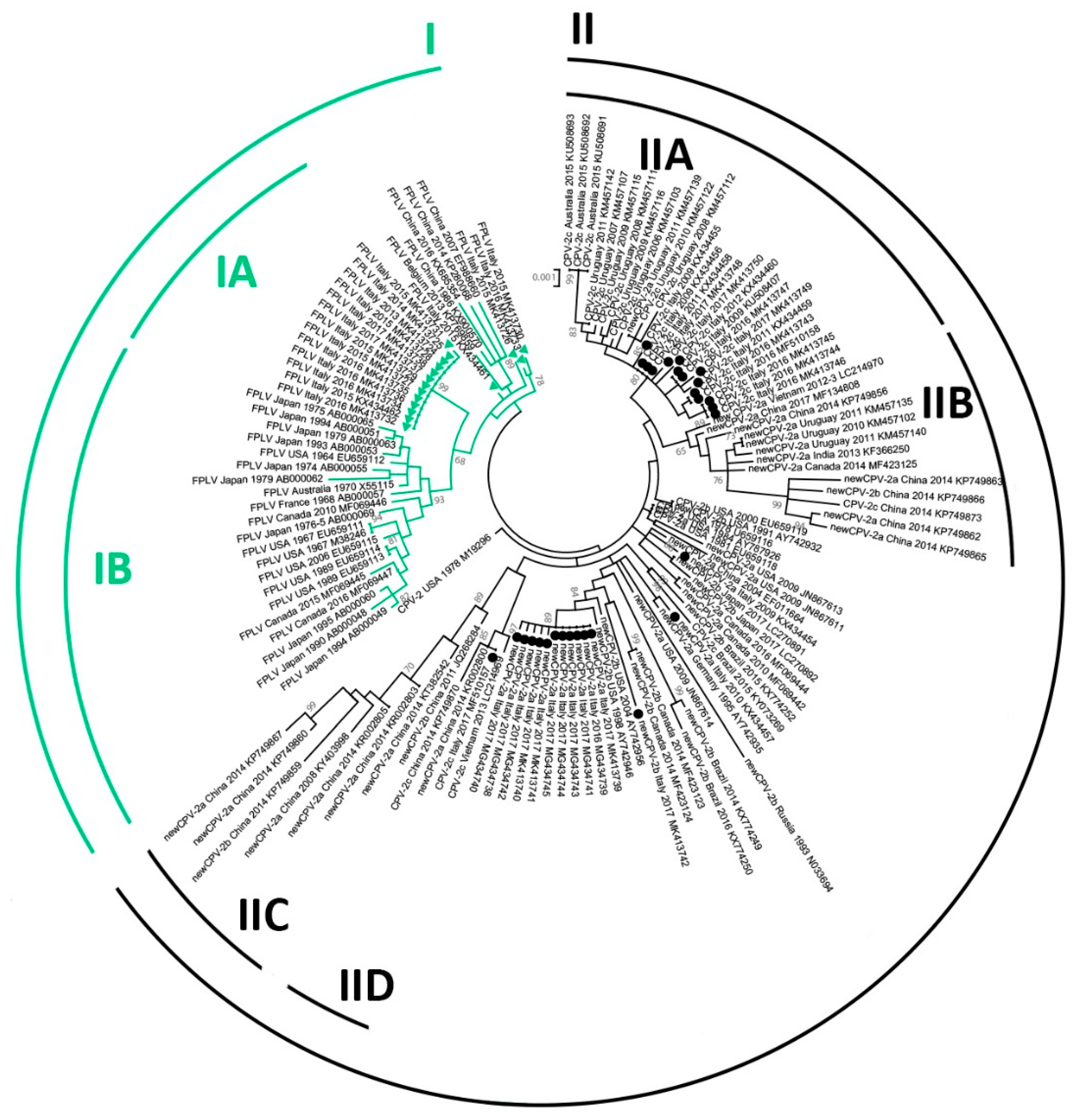
3.5. Phylogeny
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef]
- Tijssen, P.; Agbandje-McKenna, M.; Almendral, J.M.; Bergoin, M.; Flegel, T.W.; Hedman, K.; Kleinschmidt, J.; Li, Y.; Pintel, D.J.; Tattersall, P. The family Parvoviridae. In Virus Taxonomy—Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier/Academic Press: London, UK, 2011; pp. 405–425. [Google Scholar]
- Decaro, N.; Buonavoglia, C. Canine parvovirus—A review of epidemiological and diagnostic aspects, with emphasis on type 2c. Vet. Microbiol. 2012, 155, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Verge, J.; Christoforoni, N. La gastroenterite infectieuse des chats; est-elle due à un virus filtrable? CR Seances Soc. Biol. Fil. 1928, 99, 312. [Google Scholar]
- Decaro, N.; Desario, C.; Miccolupo, A.; Campolo, M.; Parisi, A.; Martella, V.; Amorisco, F.; Lucente, M.S.; Lavazza, A.; Buonavoglia, C. Genetic analysis of feline panleukopenia viruses from cats with gastroenteritis. J. Gen. Virol. 2008, 89, 2290–2298. [Google Scholar] [CrossRef]
- Truyen, U. Evolution of canine parvovirus—A need for new vaccines? Vet. Microbiol. 2006, 117, 9–13. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Parrish, C.R.; Truyen, U.; Holmes, E.C. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Natl. Acad. Sci. USA 2005, 102, 379–384. [Google Scholar] [CrossRef]
- Pereira, C.A.D.; Leal, E.S.; Durigon, E.L. Selective regimen shift and demographic growth increase associated with the emergence of high-fitness variants of canine parvovirus. Infect. Genet. Evol. 2007, 7, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Phylogenetic analysis reveals the emergence, evolution and dispersal of carnivore parvoviruses. J. Gen. Virol. 2008, 89, 2280–2289. [Google Scholar] [CrossRef]
- Decaro, N.; Desario, C.; Parisi, A.; Martella, V.; Lorusso, A.; Miccolupo, A.; Mari, V.; Colaianni, M.L.; Cavalli, A.; Di Trani, L.; et al. Genetic analysis of canine parvovirus type 2c. Virology 2009, 385, 5–10. [Google Scholar] [CrossRef]
- Parrish, C.R.; O’Connell, P.H.; Evermann, J.F.; Carmichael, L.E. Natural variation of canine parvovirus. Science 1985, 230, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Aquadro, C.F.; Strassheim, M.L.; Evermann, J.F.; Sgro, J.Y.; Mohammed, H.O. Rapid antigenic-type replacement and DNA sequence evolution of canine parvovirus. J. Virol. 1991, 65, 6544–6552. [Google Scholar]
- Buonavoglia, C.; Martella, V.; Pratelli, A.; Tempesta, M.; Cavalli, A.; Buonavoglia, D.; Bozzo, G.; Elia, G.; Decaro, N.; Carmichael, L. Evidence for evolution of canine parvovirus type 2 in Italy. J. Gen. Virol. 2001, 82, 3021–3025. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Buonavoglia, D.; Desario, C.; Amorisco, F.; Colaianni, M.L.; Parisi, A.; Terio, V.; Elia, G.; Lucente, M.S.; Cavalli, A.; et al. Characterisation of canine parvovirus strains isolated from cats with feline panleukopenia. Res. Vet. Sci. 2010, 89, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Desario, C.; Amorisco, F.; Losurdo, M.; Colaianni, M.L.; Greco, M.F.; Buonavoglia, C. Canine parvovirus type 2c infection in a kitten associated with intracranial abscess and convulsions. J. Feline Med. Surg. 2011, 13, 231–236. [Google Scholar] [CrossRef]
- Marenzoni, M.L.; Antognoni, M.T.; Baldelli, F.; Miglio, A.; Stefanetti, V.; Desario, C.; Di Summa, A.; Buonavoglia, C.; Decaro, N. Detection of parvovirus and herpesvirus DNA in the blood of feline and canine blood donors. Vet. Microbiol. 2018, 224, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, K.; Shackelton, L.A.; Holmes, E.C.; Parrish, C.R. Within-host genetic diversity of endemic and emerging parvoviruses of dogs and cats. J. Virol. 2008, 82, 11096–11105. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.P.; Jones, E.V.; Miller, T.J. Nucleotide sequence and genome organization of canine parvovirus. J. Virol. 1988, 62, 266–276. [Google Scholar]
- Hueffer, K.; Parker, J.S.L.; Weichert, W.S.; Geisel, R.E.; Sgro, J.-Y.; Parrish, C.R. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J. Virol. 2003, 77, 1718–1726. [Google Scholar] [CrossRef]
- Nelson, C.D.S.; Palermo, L.M.; Hafenstein, S.L.; Parrish, C.R. Different mechanisms of antibody-mediated neutralization of parvoviruses revealed using the Fab fragments of monoclonal antibodies. Virology 2007, 361, 283–293. [Google Scholar] [CrossRef]
- Niskanen, E.A.; Kalliolinna, O.; Ihalainen, T.O.; Häkkinen, M.; Vihinen-Ranta, M. Mutations in DNA binding and transactivation domains affect the dynamics of parvovirus NS1 protein. J. Virol. 2013, 87, 11762–11774. [Google Scholar] [CrossRef] [PubMed]
- Niskanen, E.A.; Ihalainen, T.O.; Kalliolinna, O.; Häkkinen, M.M.; Vihinen-Ranta, M. Effect of ATP binding and hydrolysis on dynamics of canine parvovirus NS1. J. Virol. 2010, 84, 5391–5403. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yuan, W.; Davis, I.; Parrish, C.R. Nonstructural protein-2 and the replication of canine parvovirus. Virology 1998, 240, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, M.; Yamaguchi, Y.; Gojobori, T.; Mochizuki, M.; Nagasawa, H.; Toyoda, Y.; Ishiguro, N.; Shinagawa, M. Differences in the evolutionary pattern of feline panleukopenia virus and canine parvovirus. Virology 1998, 249, 440–452. [Google Scholar] [CrossRef]
- Parrish, C.R. Mapping specific functions in the capsid structure of canine parvovirus and feline panleukopenia virus using infectious plasmid clones. Virology 1991, 183, 195–205. [Google Scholar] [CrossRef]
- Truyen, U.; Gruenberg, A.; Chang, S.F.; Obermaier, B.; Veijalainen, P.; Parrish, C.R. Evolution of the feline-subgroup parvoviruses and the control of canine host range in vivo. J. Virol. 1995, 69, 4702–4710. [Google Scholar]
- Pérez, R.; Calleros, L.; Marandino, A.; Sarute, N.; Iraola, G.; Grecco, S.; Blanc, H.; Vignuzzi, M.; Isakov, O.; Shomron, N.; et al. Phylogenetic and genome-wide deep-sequencing analyses of canine parvovirus reveal co-infection with field variants and emergence of a recent recombinant strain. PLoS ONE 2014, 9, e111779. [Google Scholar] [CrossRef]
- Ohshima, T.; Mochizuki, M. Evidence for recombination between feline panleukopenia virus and canine parvovirus type 2. J. Vet. Med. Sci. 2009, 71, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Y.; Liu, D.; Qiu, Z.; Tian, J.; Guo, D.; Li, Z.; Liu, M.; Li, Y.; Qu, L. Complete Genome Sequence of Feline Panleukopenia Virus Strain HRB-CS1, Isolated from a Domestic Cat in Northeastern China. Genome Announc. 2015, 3, e01556-14. [Google Scholar] [CrossRef]
- Ryt-Hansen, P.; Hagberg, E.E.; Chriél, M.; Struve, T.; Pedersen, A.G.; Larsen, L.E.; Hjulsager, C.K. Global phylogenetic analysis of contemporary aleutian mink disease viruses (AMDVs). Virol. J. 2017, 14, 231. [Google Scholar] [CrossRef]
- Mira, F.; Dowgier, G.; Purpari, G.; Vicari, D.; Di Bella, S.; Macaluso, G.; Gucciardi, F.; Randazzo, V.; Decaro, N.; Guercio, A. Molecular typing of a novel canine parvovirus type 2a mutant circulating in Italy. Infect. Genet. Evol. 2018, 61, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, T.; Liu, H.; Du, J.; Zhou, F.; Dong, Y.; He, X.; Li, Y.; Wang, C. Recombinant feline parvovirus infection of immunized tigers in central China. Emerg. Microbes Infect. 2017, 6, e42. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Britton, A.P.; Graham, S.M.; Lang, A.S. Epidemiology and molecular characterization of protoparvoviruses infecting wild raccoons (Procyon lotor) in British Columbia, Canada. Virus Res. 2017, 242, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Garigliany, M.; Gilliaux, G.; Jolly, S.; Casanova, T.; Bayrou, C.; Gommeren, K.; Fett, T.; Mauroy, A.; Lévy, E.; Cassart, D.; et al. Feline panleukopenia virus in cerebral neurons of young and adult cats. BMC Vet. Res. 2016, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Mira, F.; Purpari, G.; Lorusso, E.; Di Bella, S.; Gucciardi, F.; Desario, C.; Macaluso, G.; Decaro, N.; Guercio, A. Introduction of Asian canine parvovirus in Europe through dog importation. Transbound. Emerg. Dis. 2018, 65, 16–21. [Google Scholar] [CrossRef]
- Purpari, G.; Mira, F.; Di Bella, S.; Di Pietro, S.; Giudice, E.; Guercio, A. Investigation on canine parvovirus circulation in dogs from Sicily (Italy) by biomolecular assay. Acta Vet. (Beogr.) 2018, 68, 80–94. [Google Scholar]
- Touihri, L.; Bouzid, I.; Daoud, R.; Desario, C.; El Goulli, A.F.; Decaro, N.; Ghorbel, A.; Buonavoglia, C.; Bahloul, C. Molecular characterization of canine parvovirus-2 variants circulating in Tunisia. Virus Genes 2009, 38, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. In Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Martella, V.; Decaro, N.; Elia, G.; Buonavoglia, C. Surveillance activity for canine parvovirus in Italy. J. Vet. Med. B Infect. Dis. Vet. Public Health 2005, 52, 312–315. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Canuti, M.; O’Leary, K.E.; Hunter, B.D.; Spearman, G.; Ojkic, D.; Whitney, H.G.; Lang, A.S. Driving forces behind the evolution of the Aleutian mink disease parvovirus in the context of intensive farming. Virus Evol. 2016, 2, vew004. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Martella, V.; Elia, G.; Desario, C.; Campolo, M.; Lorusso, E.; Colaianni, M.L.; Lorusso, A.; Buonavoglia, C. Tissue distribution of the antigenic variants of canine parvovirus type 2 in dogs. Vet. Microbiol. 2007, 121, 39–44. [Google Scholar] [CrossRef]
- Shriner, D.; Nickle, D.C.; Jensen, M.A.; Mullins, J.I. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet. Res. 2003, 81, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Palermo, L.M.; Hafenstein, S.L.; Parrish, C.R. Purified feline and canine transferrin receptors reveal complex interactions with the capsids of canine and feline parvoviruses that correspond to their host ranges. J. Virol. 2006, 80, 8482–8492. [Google Scholar] [CrossRef]
- Truyen, U. Emergence and recent evolution of canine parvovirus. Vet. Microbiol. 1999, 69, 47–50. [Google Scholar] [CrossRef]
- Parker, J.S.; Parrish, C.R. Canine parvovirus host range is determined by the specific conformation of an additional region of the capsid. J. Virol. 1997, 71, 9214–9222. [Google Scholar] [PubMed]
- Truyen, U.; Evermann, J.F.; Vieler, E.; Parrish, C.R. Evolution of canine parvovirus involved loss and gain of feline host range. Virology 1996, 215, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Strassheim, M.L.; Gruenberg, A.; Veijalainen, P.; Sgro, J.Y.; Parrish, C.R. Two dominant neutralizing antigenic determinants of canine parvovirus are found on the threefold spike of the virus capsid. Virology 1994, 198, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Battilani, M.; Balboni, A.; Ustulin, M.; Giunti, M.; Scagliarini, A.; Prosperi, S. Genetic complexity and multiple infections with more Parvovirus species in naturally infected cats. Vet. Res. 2011, 42, 43. [Google Scholar] [CrossRef]
- Zhou, P.; Zeng, W.; Zhang, X.; Li, S. The genetic evolution of canine parvovirus—A new perspective. PLoS ONE 2017, 12, e0175035. [Google Scholar] [CrossRef] [PubMed]
- Nüesch, J.P.; Christensen, J.; Rommelaere, J. Initiation of minute virus of mice DNA replication is regulated at the level of origin unwinding by atypical protein kinase C phosphorylation of NS1. J. Virol. 2001, 75, 5730–5739. [Google Scholar] [CrossRef]
- Ohshima, T.; Iwama, M.; Ueno, Y.; Sugiyama, F.; Nakajima, T.; Fukamizu, A.; Yagami, K. Induction of apoptosis in vitro and in vivo by H-1 parvovirus infection. J. Gen. Virol. 1998, 79 (Pt 12), 3067–3071. [Google Scholar] [CrossRef]
- Martyn, J.C.; Davidson, B.E.; Studdert, M.J. Nucleotide sequence of feline panleukopenia virus: Comparison with canine parvovirus identifies host-specific differences. J. Gen. Virol. 1990, 71 (Pt 11), 2747–2753. [Google Scholar] [CrossRef]
- Clegg, S.R.; Coyne, K.P.; Dawson, S.; Spibey, N.; Gaskell, R.M.; Radford, A.D. Canine parvovirus in asymptomatic feline carriers. Vet. Microbiol. 2012, 157, 78–85. [Google Scholar] [CrossRef]
- Balboni, A.; Bassi, F.; De Arcangeli, S.; Zobba, R.; Dedola, C.; Alberti, A.; Battilani, M. Molecular analysis of carnivore Protoparvovirus detected in white blood cells of naturally infected cats. BMC Vet. Res. 2018, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Pommié, C.; Levadoux, S.; Sabatier, R.; Lefranc, G.; Lefranc, M.-P. IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J. Mol. Recognit. 2004, 17, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Ihalainen, T.O.; Niskanen, E.A.; Jylhävä, J.; Turpeinen, T.; Rinne, J.; Timonen, J.; Vihinen-Ranta, M. Dynamics and interactions of parvoviral NS1 protein in the nucleus. Cell. Microbiol. 2007, 9, 1946–1959. [Google Scholar] [CrossRef]
- Ihalainen, T.O.; Willman, S.F.; Niskanen, E.A.; Paloheimo, O.; Smolander, H.; Laurila, J.P.; Kaikkonen, M.U.; Vihinen-Ranta, M. Distribution and dynamics of transcription-associated proteins during parvovirus infection. J. Virol. 2012, 86, 13779–13784. [Google Scholar] [CrossRef] [PubMed]
- Mäntylä, E.; Salokas, K.; Oittinen, M.; Aho, V.; Mäntysaari, P.; Palmujoki, L.; Kalliolinna, O.; Ihalainen, T.O.; Niskanen, E.A.; Timonen, J.; et al. Promoter-Targeted Histone Acetylation of Chromatinized Parvoviral Genome Is Essential for the Progress of Infection. J. Virol. 2016, 90, 4059–4066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Mao, Y.; Xi, J.; Yu, Y.; Liu, W. Induction and suppression of type I interferon responses by mink enteritis virus in CRFK cells. Vet. Microbiol. 2017, 199, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Williams, C.V.; Gadi, S.R.; Jebbink, M.F.; Oude Munnink, B.B.; Jazaeri Farsani, S.M.; Cullen, J.M.; van der Hoek, L. Persistent viremia by a novel parvovirus in a slow loris (Nycticebus coucang) with diffuse histiocytic sarcoma. Front. Microbiol. 2014, 5, 655. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Cirone, F.; Elia, G.; Lorusso, E.; Decaro, N.; Campolo, M.; Desario, C.; Lucente, M.S.; Bellacicco, A.L.; Blixenkrone-Møller, M.; et al. Heterogeneity within the hemagglutinin genes of canine distemper virus (CDV) strains detected in Italy. Vet. Microbiol. 2006, 116, 301–309. [Google Scholar] [CrossRef]
- Decaro, N.; Campolo, M.; Elia, G.; Buonavoglia, D.; Colaianni, M.L.; Lorusso, A.; Mari, V.; Buonavoglia, C. Infectious canine hepatitis: An “old” disease reemerging in Italy. Res. Vet. Sci. 2007, 83, 269–273. [Google Scholar] [CrossRef]
- Mira, F.; Purpari, G.; Di Bella, S.; Vicari, D.; Schirò, G.; Di Marco, P.; Macaluso, G.; Battilani, M.; Guercio, A. Update on canine distemper virus (CDV) strains of Arctic-like lineage detected in dogs in Italy. Vet. Ital. 2018, 54, 225–236. [Google Scholar] [PubMed]
- Organtini, L.J.; Allison, A.B.; Lukk, T.; Parrish, C.R.; Hafenstein, S. Global displacement of canine parvovirus by a host-adapted variant: Structural comparison between pandemic viruses with distinct host ranges. J. Virol. 2015, 89, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Grecco, S.; Iraola, G.; Decaro, N.; Alfieri, A.; Alfieri, A.; Gallo Calderón, M.; da Silva, A.P.; Name, D.; Aldaz, J.; Calleros, L.; et al. Inter- and intracontinental migrations and local differentiation have shaped the contemporary epidemiological landscape of canine parvovirus in South America. Virus Evol. 2018, 4, vey011. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Sample Id * | Date of Sampling | Origin | Sample | Type | Accession Number | Reference |
|---|---|---|---|---|---|---|
| 72752/13 | 23 Oct 2013 | Cat | Spleen | FPLV | MK413724 a | This study |
| 4311/14 | 21 Jan 2014 | Cat | Spleen | FPLV | MK413725 a | This study |
| 149/15 | 05 Jan 2015 | Cat | Intestine | FPLV | MK413726 a | This study |
| 3201c1/15 | 24 Nov 2015 | Cat | Intestine | FPLV | KX434461 a | This study |
| 38056c2/15 | 26 Aug 2015 | Cat | Intestine | FPLV | MK413727 a | This study |
| 32369/15 | 14 Jul 2015 | Cat | Intestine | FPLV | MK413728 a | This study |
| 42807/15 | 24 Nov 2015 | Cat | Rectal swab | FPLV | KX434462 a | This study |
| 52333eva/15 | 18 Nov 2015 | Cat | Heart | FPLV | MK413729 a | This study |
| 55611/15 | 07 Dec 2015 | Cat | Intestine | FPLV | MK413730 a | This study |
| 58774/15 | 23 Dec 2015 | Cat | Intestine | FPLV | MK413731 a | This study |
| PA285c2/16 | 12 Jan 2016 | Cat | Intestine | FPLV | MK413732 b | This study |
| RG21/16 | 04 Jan 2016 | Cat | Spleen | FPLV | MK413733 b | This study |
| PA12880Fe/16 | 11 Apr 2016 | Cat | Intestine | FPLV | MK413734 b | This study |
| PA12880Re/16 | 11 Apr 2016 | Cat | Spleen | FPLV | MK413735 b | This study |
| PA12880Mi/16 | 11 Apr 2016 | Cat | Intestine | FPLV | MK413736 b | This study |
| PA11334/17 | 20 Apr 2017 | Cat | Brain | FPLV | MK413737 a | This study |
| CT1375/17 | 20 Feb 2017 | Cat | Spleen | FPLV | MK413738 a | This study |
| 29451/09 | 10 Sep 2009 | Dog | Intestine | CPV-2a | KX434454 a | This study |
| 987/10 | 14 Jul 2010 | Dog | Intestine | CPV-2a | KX434457 a | This study |
| PA40697/16 | 02 Nov 2016 | Dog | Spleen | CPV-2a | MK413739 b | This study |
| PA43847/16 | 21 Nov 2016 | Dog | Rectal swab | CPV-2a | MG434738 a | [32] |
| PA48686/16 | 21 Dec 2016 | Dog | Intestine | CPV-2a | MG434739 a | [32] |
| PA3213/17 | 09 Feb 2017 | Dog | Intestine | CPV-2a | MG434740 a | [32] |
| PA5610/17 | 03 Mar 2017 | Dog | Rectal swab | CPV-2a | MG434741 a | [32] |
| PA10388/17 | 11 Apr 2017 | Dog | Spleen | CPV-2a | MG434742 a | [32] |
| PA13577/17 | 15 May 2017 | Dog | Spleen | CPV-2a | MG434743 a | [32] |
| PA13579id90/17 | 15 May 2017 | Dog | Intestine | CPV-2a | MG434744 a | [32] |
| PA13579id93/17 | 15 May 2017 | Dog | Spleen | CPV-2a | MG434745 a | [32] |
| PA30636/17 | 31 Oct 2017 | Dog | Spleen | CPV-2a | MK413740 a | This study |
| PA31209/17 | 07 Nov 2017 | Dog | Spleen | CPV-2a | MK413741 a | This study |
| PA13600/17 | 15 May 2017 | Dog | Spleen | CPV-2b | MK413742 a | This study |
| 23782/09 | 10 Sep 2009 | Dog | Intestine | CPV-2c | KX434455 a | This study |
| 25835/09 | 10 Sep 2009 | Dog | Intestine | CPV-2c | KU508407 a | This study |
| 45361/09 | 21 Oct 2009 | Dog | Intestine | CPV-2c | KX434456 a | This study |
| 2323/11 | 21 Jun 2011 | Dog | Intestine | CPV-2c | KX434458 a | This study |
| 27692c1/11 | 05 Jul 2011 | Dog | Intestine | CPV-2c | KX434459 a | This study |
| 52238/12 | 20 Oct 2012 | Dog | Intestine | CPV-2c | KX434460 a | This study |
| PA15423/16 | 29 Apr 2016 | Cat | Spleen | CPV-2c | MK413743 a | This study |
| PA36395/16 | 06 Oct 2016 | Dog | Intestine | CPV-2c | MK413744 a | This study |
| PA39667/16 | 26 Oct 2016 | Dog | Brain | CPV-2c | MK413745 b | This study |
| 41113c1/16 | 03 Nov 2016 | Dog | Rectal swab | CPV-2c | MF510158 a | [36] |
| PA41113c2/16 | 03 Nov 2016 | Dog | Rectal swab | CPV-2c | MK413746 a | This study |
| PA45984/16 | 01 Dec 2016 | Dog | Rectal swab | CPV-2c | MK413747 a | This study |
| 2743/17 | 06 Feb 2017 | Dog | Intestine | CPV-2c | MF510157 a | [36] |
| CT1839id0018/17 | 02 Mar 2017 | Dog | Intestine | CPV-2c | MK413748 a | This study |
| CT1839id2213/17 | 02 Mar 2017 | Dog | Intestine | CPV-2c | MK413749 a | This study |
| PA27184/17 | 29 Sep 2017 | Dog | Rectal swab | CPV-2c | MK413750 a | This study |
| Strain | NS1 Amino Acids (Nucleotides) a | ||||||
|---|---|---|---|---|---|---|---|
| 81 (241–243) | 115 (343–345) | 247 (739–741) | 248 (742–744) | 545 (1633–1635) | 595 (1783–1785) | 664 (1990–1992) | |
| 72752/13 | V (GTT) | I (ATT) | H (CAT) | T (ACT) | E (GAA) | H (CAC) | Q (CAA) |
| 4311/14 | -- | -- | -- | -- | -- | -- | -- |
| 149/15 | I (ATT) | V (GTT) | Q (CAA) | -- | -- | Q (CAA) | R (CGA) |
| 3201c1/15 | -- | V (GTT) | Q (CAA) | -- | -- | Q (CAA) | R (CGA) |
| 55611/15 | -- | V (GTT) | Q (CAA) | -- | -- | Q (CAA) | R (CGA) |
| RG21/16 | -- | V (GTT) | Q (CAA) | -- | -- | Q (CAA) | R (CGA) |
| 38056c/15 | -- | -- | -- | -- | -- | -- | -- |
| 32369/15 | -- | -- | -- | -- | -- | -- | -- |
| 42807/15 | -- | -- | -- | -- | -- | -- | -- |
| 52333eva/15 | -- | -- | -- | -- | -- | -- | -- |
| 58774/15 | -- | -- | -- | -- | -- | -- | -- |
| PA285c2/16 | -- | -- | -- | -- | -- | -- | -- |
| PA12880Felix/16 | -- | -- | -- | -- | -- | -- | -- |
| PA12880Red/16 | -- | -- | -- | -- | -- | -- | -- |
| PA12880Miele/16 | -- | -- | -- | -- | -- | -- | -- |
| PA11334/17 | -- | -- | -- | -- | -- | -- | -- |
| CT1375/17 | -- | -- | -- | -- | -- | -- | -- |
| Strain | NS1 Amino Acids (Nucleotides) b | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CPV Variant | 60 (178–180) | 239 (715–717) | 247 (739–741) | 248 (742–744) | 350 (1048–1050) | 397 (1189–1191) | 544 (1630–1632) | 545 (1633–1635) | 572 (1714–1716) | 584 (1750–1752) | 590 (1768–1770) | 595 (1783–1785) | 597 (1789–1791) | 630 (1888–1890) | |
| 29451/09 | CPV-2a | I (ATT) | N (AAC) | Q (CAA) | I (ATT) | D (GAT) | L (CTT) | Y (TAT) | E (GAA) | K (AAA) | T (ACA) | P (CCT) | Q (CAA) | L (CTA) | L (CTT) |
| 987/10 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | A (GCA) | -- | -- | -- | -- |
| PA40697/16 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA43847/16 | CPV-2a | -- | T (ACC) | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA48686/16 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA3213/17 | CPV-2a | -- | -- | -- | -- | N (AAT) | F (TTT) | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA5610/17 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA10388/17 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA13577/17 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA13579id90/17 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA13579id93/17 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA30636/17 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA31209/17 | CPV-2a | -- | -- | -- | -- | N (AAT) | -- | F (TTT) | -- | E (GAA) | -- | -- | -- | P (CCA) | -- |
| PA13600/17 | CPV-2b | -- | -- | -- | -- | -- (AAC) | -- | -- | -- | -- | -- | S (TCT) | -- | P (CCA) | -- |
| 23782/09 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| 25835/09 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| 45361/09 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| 2323/11 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| 27692c1/11 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| 52238/12 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| PA15423/16 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| PA36395/16 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| PA39667/16 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| 41113c1/16 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| PA41113c2/16 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| PA45984/16 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| 2743/17 | CPV-2c | V (GTT) | -- | -- | -- | -- | -- | F (TTT) | V (GTA) | E (GAA) | -- | -- | -- | -- | P (CCT) |
| CT1839id0018/17 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| CT1839id2213/17 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| PA27184/17 | CPV-2c | -- | -- | -- | -- | -- | -- | -- | -- | E (GAA) | -- (ACG) | -- | -- | -- | -- |
| Sites Under Negative Selection Pressure * | Sites Under Positive Selection Pressure | ||
|---|---|---|---|
| FUBAR | FUBAR | MEME | |
| CPV | 7, 10, 14, 15, 31, 32, 47, 53, 54, 56, 66, 68, 69, 83, 92, 99, 102, 104, 105, 107, 114, 119, 123, 124, 132, 135, 137, 140, 154, 163, 164, 165, 170, 172, 179, 189, 200, 211, 219, 223, 240, 242, 250, 251, 279, 283, 284, 297, 307, 313, 323, 324, 325, 333, 336, 337, 340, 341, 343, 349, 353, 360, 366, 371, 374, 378, 384, 388, 391, 393, 394, 395, 403, 405, 408, 430, 432, 435, 439, 444, 451, 459, 463, 467, 473, 474, 475, 476, 483, 488, 489, 494, 495, 497, 499, 503, 505, 506, 512, 514, 517, 525, 527, 528, 529, 531, 536, 537, 541, 543, 554, 560, 563, 564, 584, 591, 596, 633, 640, 641, 642, 657, 659, 662 | 19, 278, 545, 572, 583 | 278, 572, 583, 597, 647 |
| FPLV | 6, 31, 39, 58, 60, 71, 97, 102, 174, 177, 185, 201, 207, 270, 284, 307, 323, 352, 357, 403, 418, 422, 428, 435, 462, 479, 488, 489, 493, 515, 517, 520, 533, 540, 541, 543, 549, 551, 560, 562, 653, 660 | 443 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mira, F.; Canuti, M.; Purpari, G.; Cannella, V.; Di Bella, S.; Occhiogrosso, L.; Schirò, G.; Chiaramonte, G.; Barreca, S.; Pisano, P.; et al. Molecular Characterization and Evolutionary Analyses of Carnivore Protoparvovirus 1 NS1 Gene. Viruses 2019, 11, 308. https://doi.org/10.3390/v11040308
Mira F, Canuti M, Purpari G, Cannella V, Di Bella S, Occhiogrosso L, Schirò G, Chiaramonte G, Barreca S, Pisano P, et al. Molecular Characterization and Evolutionary Analyses of Carnivore Protoparvovirus 1 NS1 Gene. Viruses. 2019; 11(4):308. https://doi.org/10.3390/v11040308
Chicago/Turabian StyleMira, Francesco, Marta Canuti, Giuseppa Purpari, Vincenza Cannella, Santina Di Bella, Leonardo Occhiogrosso, Giorgia Schirò, Gabriele Chiaramonte, Santino Barreca, Patrizia Pisano, and et al. 2019. "Molecular Characterization and Evolutionary Analyses of Carnivore Protoparvovirus 1 NS1 Gene" Viruses 11, no. 4: 308. https://doi.org/10.3390/v11040308