1. Introduction
Virus infections present serious health threats to millions of individuals worldwide. For public health, the accurate detection of pathogenic viruses is time-critical because reducing the time for diagnosis and treatment lowers the risk of disease transmission and patient mortality. Fast and robust diagnostic assays are therefore required to rapidly detect re-emerging and newly emerging viruses (e.g., Influenza, Ebola, Zika, or Hepatitis C virus). These diagnostic methods need to cover a broad spectrum of potentially disease-causing viral agents.
Classical diagnostic strategies for detecting viral infection can be divided into two different categories: on the one hand, virus detection can be established by targeted methods, such as agent-specific polymerase chain reaction (PCR) or immunological techniques. On the other hand, detection approaches exist that provide an open view, such as electron microscopy or next-generation sequencing (NGS). Besides their unbiased view, the latter methods have the advantage of identifying multiple pathogens in a single experimental run. Due to its specificity (hybridization and sequencing) and sensitivity (qPCR), the detection of nucleic acids is the gold standard in diagnostics. Conversely, the detection of viral proteins is used less frequently in diagnostic settings and is usually based on interaction with affine binding molecules such as antibodies or aptamers. However, producing these binding molecules is generally time-consuming and laborious, as is the validation of their specificity.
While in clinical microbiology the analysis of subproteomes (<12 kDa) using matrix assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry (MS) has become a standard method for the identification of bacteria and fungi, no comparable proteomic approach exists in virology for technical reasons [
1]. In recent years, MS-based targeted proteomics has evolved into a technique for detecting proteins in complex samples with high sensitivity, quantitative accuracy, and reproducibility [
2,
3]. Targeted proteomics is commonly used to test hypotheses on a subset of proteins of interest, in contrast to discovery shotgun proteomics. The latter provides global proteome profiling of thousands of proteins in a sample, however, at the expense of sensitivity and reproducibility. Unlike discovery methods, targeted methods of selected/multiple reaction monitoring (SRM/MRM) [
4] and parallel reaction monitoring (PRM) [
5] nowadays allow for detecting and analyzing preselected proteins and peptides in sensitive, specific, and time-efficient manner. Furthermore, the development of targeted proteomics assays has become easier in the past few years, owing to the advances of analytical methods, instrumental capabilities, and computational workflows [
6].
Targeted MS-based proteomics assay development typically involves (i) peptide candidate selection, (ii) peptide synthesis, and (iii) assay optimization. This procedure now enables the transfer of a process highly similar to the design of multiplex PCRs to the proteome level for detecting pathogens. While MS-based targeted assays have not been used for detecting viruses in any diagnostic setting yet, promising findings could already be achieved for identifying and quantifying pathogenic bacterial species. For example, targeted proteomics methods were successfully used in previous studies on
Streptococcus pyogenes [
7] and
Mycobacterium tuberculosis [
8].
Although targeted proteomics has gained much popularity with many use cases in experimental research by now, relatively few research-oriented algorithms and software tools have been developed that support the user-defined selection of peptides for designing targeted SRM or PRM assays. In this context, Skyline [
9] is a powerful and widely used software for designing targeted proteomics assays. Besides its wide applicability to different targeted methods and its intuitive use, it also has some internal limitations: first, Skyline is dependent on the operating system Windows, and can therefore not be used under a Linux cluster server environment, and second, it does perform only exact string matching during the peptide selection process without considering any homologies between related organisms. PeptidePicker [
10] is a web-based workflow to select peptides by providing, amongst further options, the protein accession number and was designed for human and mouse proteomes. PeptideManager [
11] is a tool developed to select peptide candidates as protein surrogates from a defined proteome. It was optimized for the use case of xenografts, i.e., human tumors orthotopically implanted into a different species. While this software allows for constructing a peptide database from any species-specific proteome, sequence homologies, and multiple taxonomic levels are disregarded. Picky [
12]—a web-based method designer for targeted assays—only provides support for human and mouse sequences, while it relies on synthetic peptide data from the human-focused ProteomeTools project [
13,
14]. In the context of targeted metaproteomics, the Unique Peptide Finder of the UniPept web application [
15] was developed to select unique peptides for user-defined taxa. Furthermore, various computational tools have been developed to predict proteotypic peptides for targeted proteomics experiments [
16,
17,
18]. These methods often make use of machine learning training setups that incorporate the probability of observing a peptide in a standard proteomics analysis, referred to as peptide detectability [
19] or observability [
20], and commonly involve physicochemical properties of the proteins to select high-responding peptides [
21]. To our best knowledge, however, no software tool is currently available to select taxon-specific peptides for targeted proteomics assays that also accounts for sequence homologies between different species or strain proteomes. Effectively considering homologies is crucial for accurate taxon-specific diagnostics, because proteins measured in virus samples frequently have a high sequence similarity either in closely related strains or due to highly conserved functional domains.
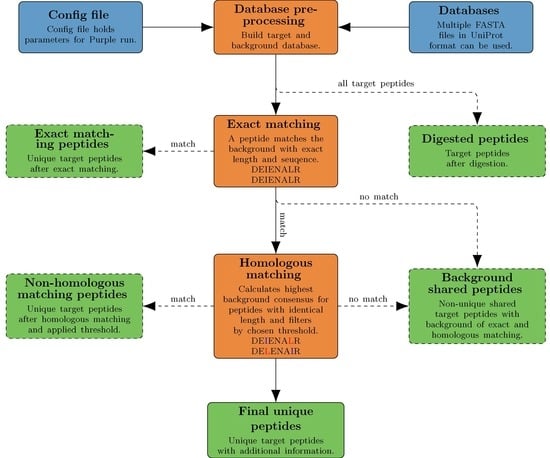
Here we present Purple (Picking unique relevant peptides for viral experiments), a platform-independent software that returns a set of taxon-specific peptides, after the user has specified the viral target (i.e., a particular virus species or genus), as candidates for targeted proteomics experiments. Equipped with a user-friendly graphical user interface and a threshold-based filtering strategy for homologous sequences, it simplifies the design of MS-based targeted proteomics assays for the end user. Purple enables peptide candidate selection and considers background sequence information, i.e., proteins that are not related to a specific virus target, at various taxonomic levels. Thus, all peptide candidates are validated against a user-defined database of virus proteomes. While the design of MS-based targeted assays requires further steps, our software greatly facilitates the cumbersome, yet important task of peptide selection and thereby paves the way to time-efficient and robust pathogen screening and viral diagnostics. Purple is open source software available at
https://gitlab.com/rki_bioinformatics/Purple.
4. Discussion
The main goal of our developed Purple software is to provide taxon-specific peptides for a targeted proteomics assay. These targeted assays can be used in a diagnostic setting to identify a virus species/strain or even a whole virus family in a sample in sensitive and time-efficient manner. In this work, we validated the software in three different benchmarking experiments.
Purple enabled us to retrieve taxon-specific peptides to distinguish between arenavirus species proteomes that are very similar in their sequences (see
Section 3.1). Accordingly, we observed a comparable decrease in the ratio of unique to
in silico-digested peptides for New and Old World arenaviruses based on differences between their proteomes (
Figure 4). This effect could also be recognized also on the clade level for the New World viruses.
The data analysis of CPXV (see
Section 3.2) resulted in 56 taxon-specific peptides (
Figure 7). These peptides were present in each MS/MS sample replicate and can be used to uniquely identify CPXV in a mixed biological sample, although its proteome is very similar to other Orthopoxvirus species and strains (
Figure 8). By changing to a Brighton Red strain-specific target database, a reliable determination of the strain would be possible as well. This underlines that Purple relies on a correct and complete database to yield to the best possible results. Missing or incorrectly assigned protein sequences could result in incorrect selected unique peptides or discarded ones. Furthermore, although many spectra in the shotgun proteomics experiment were assigned to human peptides, this does not present a limitation for the targeted proteomics approach, because unique virus peptides selected by Purple can be detected using a targeted (e.g., PRM-based) assay in specific and sensitive manner; for example, in a recently published study [
35], a PRM-based assay was used to identify dengue virus species directly from clinical serum samples. Nevertheless, to validate the resulting set of peptides, it would be recommended to test them on other CPXV samples and to check if the peptides are detectable in these samples likewise. In addition, the selected background database might be incomplete, e.g., when proteome references were missed to be included for the Purple analysis. In this case, it is useful to validate Purple-selected peptides using secondary tools such as Unipept [
36] for resolving the taxonomic origin of any tryptic peptide based on the complete UniProt database. Furthermore, false negatives may result from issues during sample preparation or poor instrument performance. Therefore, these parameters need to be controlled in diagnostic PRM assays, e.g., by using internal standards and running further quality control samples.
It can be crucial in virus infection scenarios to accurately distinguish between specific strains. To cover these cases, we examined the strain-level resolution of our tool using data of VACV Copenhagen and VACV Western Reserve strains (see
Section 3.3). Purple was able to find a reliable amount of strain-specific peptides (
Table 7). The intersection between the Purple-selected peptides and the peptide identification from the database search showed that it is possible to detect these peptides. In general, strain-level identification was possible even for an applied FDR threshold of 1%, however, it became apparent that the shotgun proteomics approach becomes limited due to the spurious numbers of identified peptides. The number of peptides could be increased by adjusting the FDR filtering or by using a targeted proteomics approach with higher sensitivity.
In comparison to other tools, Purple offers several advantages, such as cross-platform compatibility on multiple operating systems. Purple allows a homology-based analysis of multiple proteome databases at once and produces an aggregated and summarized export on various levels. In addition, Purple is not limited to specific organisms, but can be used with general UniProt databases, also including eukaryotic and bacterial databases. High sequence similarity between strains and horizontal gene transfer may complicate taxon-specific classification for bacterial samples. However, Purple could help to overcome complications and can be helpful for creating targeted assays for bacterial detection as well. The graphical user interface and compatibility with all UniProt databases enables researchers without bioinformatics background to find taxon-specific peptides in an easy and straightforward manner.
A potential improvement to the software would be to move from a sequence identity-based metric based on the Hamming distance to similarity-based matching for the homologous matching mode. In this case, amino acid substitutions are not weighted equally, for example by using a PAM or BLOSUM matrix [
37]. This similarity-based metric might allow a more accurate homologous matching in Purple. For example, an approach based on a structural alignment as introduced by Ogata et al. [
38] might be useful. Further potential improvements with useful features in Purple include adding plots for better data exploration and a tabular view for inspecting the results (that are currently exportable as text files to spreadsheet software).
In summary, the most promising application of Purple is to select taxon-specific peptides for creating tailored SRM or PRM assays with high sensitivity and specificity. This application will allow for new time- and cost-efficient diagnostic methods in healthcare and further biological applications. It could even be used to identify multiple organisms in a single sample in the context of targeted metaproteomics [
39].

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}