Phages and Human Health: More Than Idle Hitchhikers


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Phage Biology
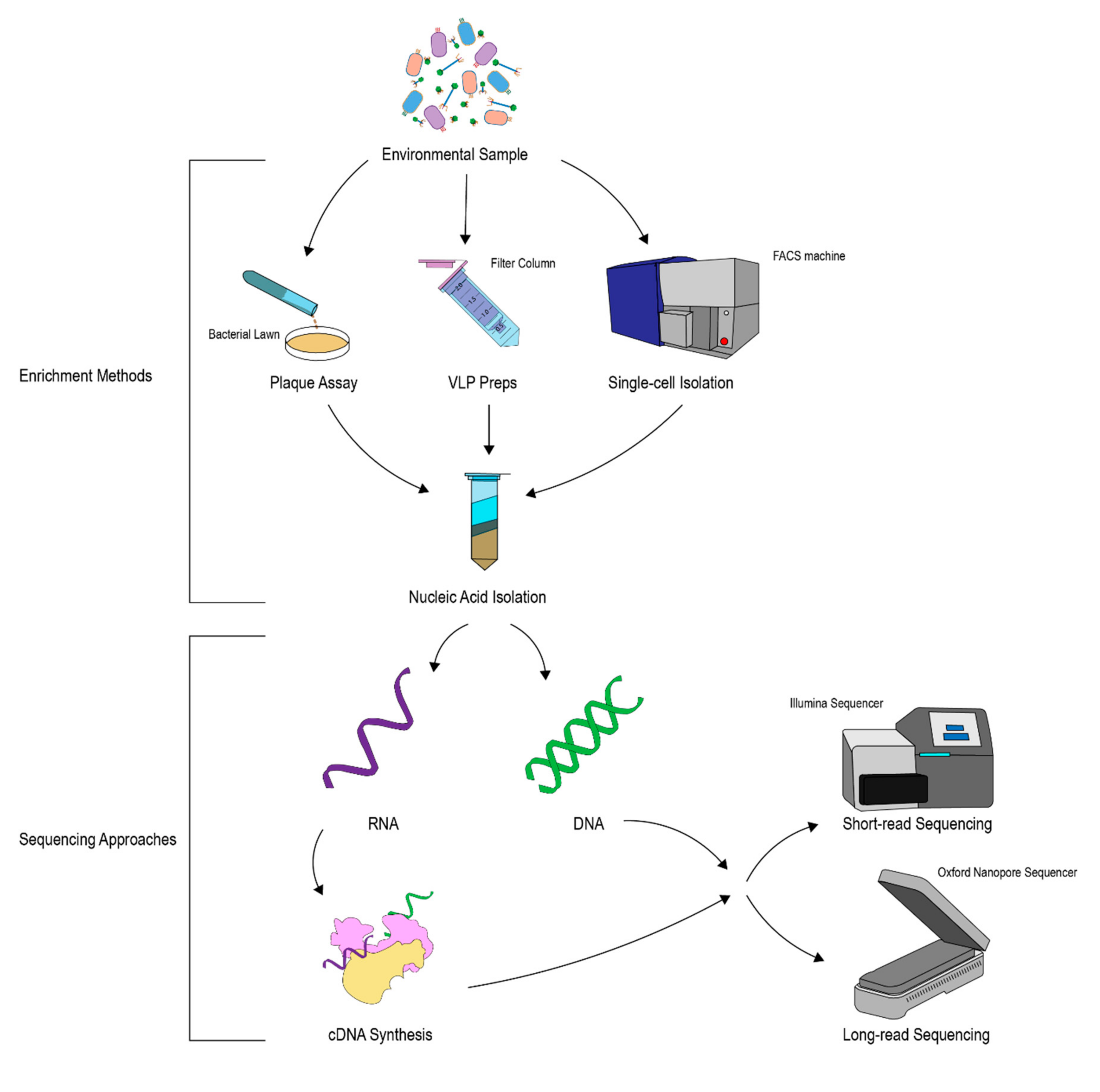
3. Isolation and Genomic Analysis of Phages
3.1. Isolation of Phages
3.2. Computational Identification of Phages
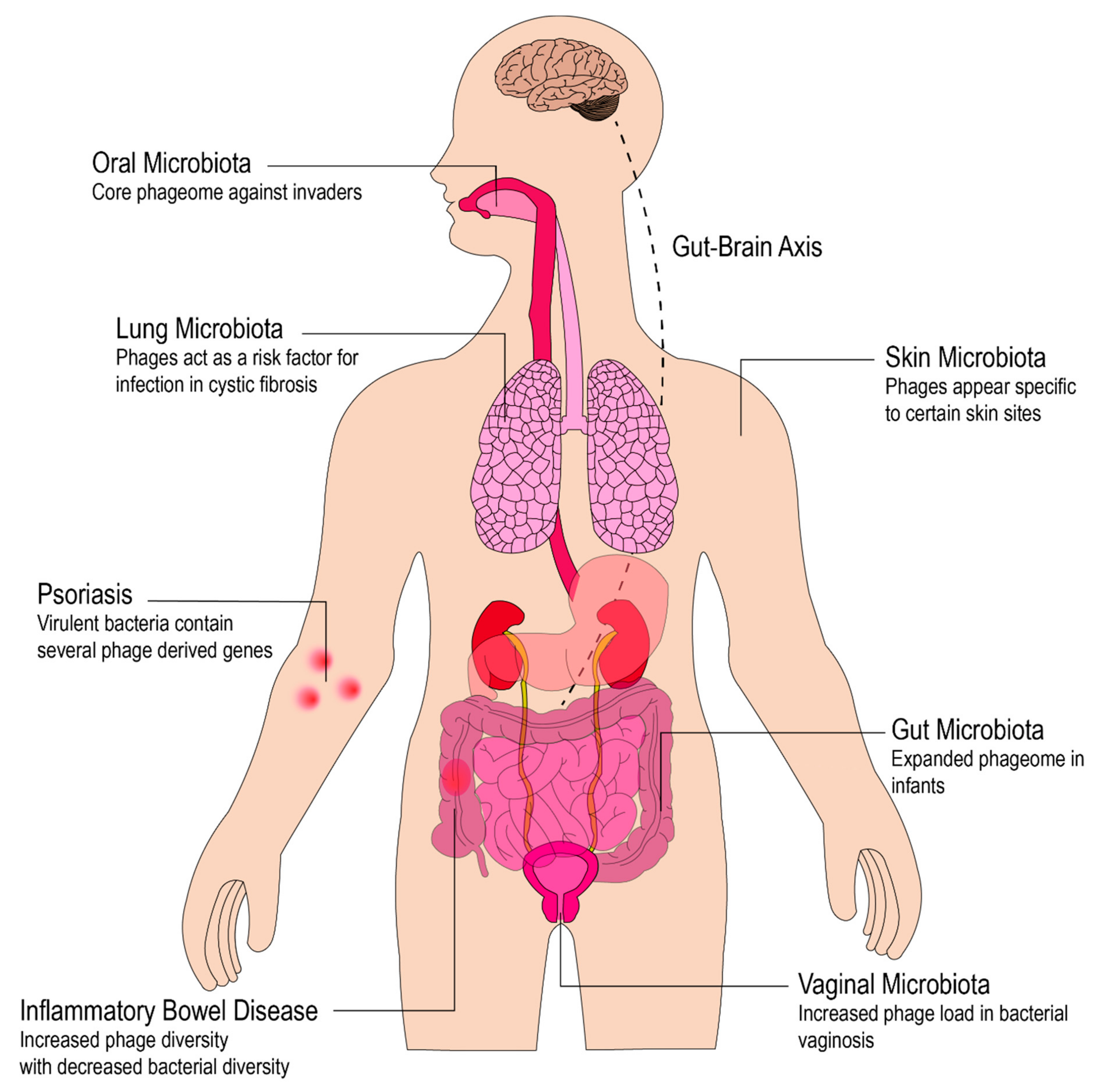
4. The Role of Phages in the Human Microbiota
4.1. Phages in the Infant Gut
4.2. Phages in the Adult Gut
4.3. Phages on the Skin
4.4. Phages in the Vaginal Microbiota
4.5. Phages in the Lungs
5. The Future of Phages in Research and Health
Acknowledgments
Conflicts of Interest
References
- Twort, F. An investigation on the nature of ultra-microscopic viruses. Lancet 1915, 186, 1241–1243. [Google Scholar] [CrossRef]
- Herelle, F.D. An invisible microbe that is antagonistic to the dysentery bacillus Cozzes rendus. Acad. Sci. 1917, 165, 373–375. [Google Scholar]
- Ackermann, H. Bacteriophage observations and evolution. Res. Microbiol. 2003, 154, 245–251. [Google Scholar] [CrossRef]
- Dulbecco, R.; Vogt, M. Some problems of animal virology as studied by the plaque technique. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; McNair, K.; Faust, K.; Raes, J.; Dutilh, B.E. Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev. 2016, 40, 258–272. [Google Scholar] [CrossRef]
- Jensen, E.C.; Schrader, H.S.; Rieland, B.; Thompson, T.L.; Lee, K.W.; Nickerson, K.W.; Kokjohn, T.A. Prevalence of Broad-Host-Range Lytic Bacteriophages of Sphaerotilus natans, Escherichia coli, and Pseudomonas aeruginosa. Appl. Environ. Microbiol. 1998, 64, 575–580. [Google Scholar] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.; Knight, R.; Gordon, J.I. The human microbiome project: Exploring the microbial part of ourselves in a changing world Nature. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [Green Version]
- Van Zyl, L.J.; Abrahams, Y.; Stander, E.A.; Kirby-McCollough, B.; Jourdain, R.; Clavaud, C.; Breton, L.; Trindade, M. Novel phages of healthy skin metaviromes from South Africa. Sci. Rep. 2018. [Google Scholar] [CrossRef]
- Abedon, S.T.; García, P.; Mullany, P.; Aminov, R. Editorial: Phage Therapy: Past, Present and Future. Front. Microbiol. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mertz, L. Battling Superbugs: How Phage Therapy Went From Obscure to Promising. IEEE Pulse 2019, 10, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Ofir, G.; Sorek, R. Contemporary Phage Biology: From Classic Models to New Insights. Cell 2018, 172, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutter, E.; Sulakvelidz, A. Bacteriophages: Biology and Applications; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Łobocka, M.B.; Rose, D.J.; Plunkett, G.; Rusin, M.; Samojedny, A.; Lehnherr, H.; Yarmolinsky, M.B.; Blattner, F.R. Genome of Bacteriophage P1. J. Bacteriol. 2004, 186, 7032–7068. [Google Scholar] [Green Version]
- Olszak, T.; Latka, A.; Roszniowski, B.; Valvano, M.A.; Drulis-Kawa, Z. Phage Life Cycles Behind Bacterial Biodiversity. Curr. Med. Chem. 2017, 24, 3987–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goerke, C.; Ko, J.; Wolz, C. Ciprofloxacin and Trimethoprim Cause Phage Induction and Virulence Modulation in Staphylococcus aureus. Microbiology 2006, 50, 171–177. [Google Scholar] [CrossRef]
- Diard, M.; Bakkeren, E.; Cornuault, J.K.; Moor, K.; Hausmann, A.; Sellin, M.E.; Loverdo, C.; Aertsen, A.; Ackermann, M.; De Paepe, M.; et al. Inflammation boosts bacteriophage transfer between Salmonella spp. Science 2017, 355, 1211–1215. [Google Scholar] [CrossRef]
- Hobbs, Z.; Abedon, S.T. Diversity of phage infection types and associated terminology: The problem with “Lytic or Lysogenic”. FEMS Microbiol. Lett. 2016, 363, 1–8. [Google Scholar] [CrossRef]
- Cenens, W.; Makumi, A.; Mebrhatu, M.T.; Lavigne, R.; Aertsen, A. Phage–host interactions during pseudolysogeny. Bacteriophage 2013, 3, e25029. [Google Scholar] [CrossRef]
- Abedon, S.T. Disambiguating Bacteriophage Pseudolysogeny: An Historical Analysis of Lysogeny, Pseudolysogeny, and the Phage Carrier State. Contemp. Trends Bacterioph. Res. 2009, 285–307. [Google Scholar]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Wittmann, J.; Kuhn, J.H.; Turner, D.; Sullivan, M.B.; Dutilh, B.E.; Bin Jang, H.; Van Zyl, L.J.; Klumpp, J.; Lobocka, M.; et al. Taxonomy of prokaryotic viruses: 2017 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2018, 163, 1125–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barylski, J.; Enault, F.; Dutilh, B.E.; Schuller, M.B.; Edwards, R.A.; Gillis, A.; Klumpp, J.; Knezevic, P.; Krupovic, M.; Kuhn, J.H.; et al. Taxonomy proposal To create one (1) new family, Herelleviridae, in the order Caudovirales. In ICTV Online: International Committee on Taxonomy of Viruses (ICTV); International Committee on Taxonomy of Viruses (ICTV), 2018; p. 2018.118B. [Google Scholar]
- López-Pérez, M.; Haro-Moreno, J.M.; Gonzalez-Serrano, R.; Parras-Moltó, M.; Rodríguez-Valera, F. Genome diversity of marine phages recovered from Mediterranean metagenomes: Size matters. PLoS Genet. 2017, 13, e1007018. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Janowski, A.B.; Zhao, G.; Barouch, D.; Wang, D. Hyperexpansion of RNA Bacteriophage Diversity. PLoS Biol. 2016, 14, e1002409. [Google Scholar] [CrossRef] [PubMed]
- Twest, R.; Kropinski, A.M. Bacteriophage Enrichment from Water and Soil. In RNA Scaffolds; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2009; Volume 501, pp. 15–21. [Google Scholar]
- Koren, S.; Phillippy, A.M. One chromosome, one contig: Complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 2015, 23, 110–120. [Google Scholar] [CrossRef]
- Berrios, L.; Ely, B. Correction to: The Isolation and Characterization of Kronos, a Novel Caulobacter Rhizosphere Phage that is Similar to Lambdoid Phages. Curr. Microbiol. 2019, 76, 964–965. [Google Scholar] [CrossRef] [Green Version]
- Warwick-Dugdale, J.; Solonenko, N.; Moore, K.; Chittick, L.; Gregory, A.C.; Allen, M.J.; Sullivan, M.B.; Temperton, B. Long-read viral metagenomics enables capture of abundant and microdiverse viral populations and their niche-defining genomic islands. In Genomics; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2018; p. 345041. [Google Scholar]
- Humphrey, S.B.; Jensen, N.S.; Stanton, T.B. Mitomycin C induction of bacteriophages from Serpulina hyodysenteriae and Serpulina innocens. FEMS Microbiol. Lett. 1995, 134, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B.; Bishop-Lilly, K. VirSorter: Mining viral signal from microbial genomic data. Peer J. 2015, 3, 985. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, 000132. [Google Scholar] [CrossRef]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, M.; Hooper, L.V.; A Duerkop, B. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genom. 2015, 16, 1915. [Google Scholar] [CrossRef] [PubMed]
- Poulos, B.T.; John, S.G.; Sullivan, M.B. Iron Chloride Flocculation of Bacteriophages from Seawater; Humana Press: New York, NY, USA, 2018; pp. 49–57. [Google Scholar]
- Labonté, J.M.; Swan, B.K.; Poulos, B.; Luo, H.; Koren, S.; Hallam, S.J.; Sullivan, M.B.; Woyke, T.; Wommack, K.E.; Stepanauskas, R. Single-cell genomics-based analysis of virus–host interactions in marine surface bacterioplankton. ISME J. 2015, 9, 2386–2399. [Google Scholar] [CrossRef] [PubMed]
- Parikka, K.J.; Romancer, M.L.; Wauters, N. Deciphering the virus-to-prokaryote ratio (VPR): insights into virus–host relationships in a variety of ecosystems. Biol. Rev. 2017, 1083, 1081–1100. [Google Scholar] [CrossRef] [PubMed]
- Woyke, T.; Tighe, D.; Mavromatis, K.; Clum, A.; Copeland, A.; Schackwitz, W.; Lapidus, A.; Wu, D.; McCutcheon, J.P.; McDonald, B.R. One Bacterial Cell, One Complete Genome. PLoS ONE 2010, 5, e10314. [Google Scholar] [CrossRef] [PubMed]
- Martinez-hernandez, F.; Garcia-heredia, I.; Martinez-garcia, M. Deciphering the Human Virome with Single-Virus genomics and metagenomics. Viruses 2018, 10, 113. [Google Scholar]
- Marbouty, M.; Baudry, L.; Cournac, A.; Koszul, R. Scaffolding bacterial genomes and probing host-virus interactions in gut microbiome by proximity ligation (chromosome capture) assay. Sci. Adv. 2017, 3, e1602105. [Google Scholar] [CrossRef]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef] [PubMed]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Prophinder: A computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 2008, 24, 863–865. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.K.; Edwards, R.A. PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012, 40, e126. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39 (Suppl. 2), 347–352. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkman, N.E.; Villegas, E.N.; Garland, J.L.; Keely, S.P. Reducing inherent biases introduced during DNA viral metagenome analyses of municipal wastewater. PLoS ONE 2018, 13, e0195350. [Google Scholar] [CrossRef] [PubMed]
- Brister, J.R.; Ako-adjei, D.; Bao, Y.; Blinkova, O. NCBI Viral Genomes Resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef]
- Nelson, K.E.; Weinel, C.; Paulsen, I.T.; Dodson, R.J.; Hilbert, H.; Dos Santos, V.A.P.M.; Fouts, D.E.; Gill, S.R.; Pop, M.; Holmes, M.; et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 2002, 4, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Ahlgren, N.A.; Ren, J.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. Alignment-free d2∗ oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences. Nucleic Acids Res. 2017, 45, 39–53. [Google Scholar] [CrossRef]
- Pride, D.T.; Salzman, J.; Haynes, M.; Rohwer, F.; Davis-Long, C.; White, R.A., III; Loomer, P.; Armitage, G.C.; Relman, D.A. Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J. 2012, 6, 915–926. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Zhao, F. Phage-bacteria interaction network in human oral microbiome. Environ. Microbiol. 2016, 18, 2143–2158. [Google Scholar] [CrossRef]
- Gregory, A.C.; Sullivan, M.B.; Segal, L.N.; Keller, B.C. Smoking is associated with quantifiable differences in the human lung DNA virome and metabolome. Respir. Res. 2018, 19, 174. [Google Scholar] [CrossRef] [PubMed]
- Burgener, E.B.; Sweere, J.M.; Bach, M.S.; Secor, P.R.; Haddock, N.; Jennings, L.K.; Marvig, R.L.; Johansen, H.K.; Rossi, E.; Cao, X.; et al. Filamentous bacteriophages are associated with chronic Pseudomonas lung infections and antibiotic resistance in cystic fibrosis. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Byrd, A.L.; Park, M.; Kong, H.H.; Segre, J.A. Temporal Stability of the Human Skin Microbiome. Cell 2016, 165, 854–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, E.; Shi, B.; Kang, D.; Craft, N.; Li, H. The balance of metagenomic elements shapes the skin microbiome in acne and health. Sci. Rep. 2016, 6, 39491. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yan, R.; Zhong, Q.; Ngo, S.; Bangayan, N.J.; Nguyen, L.; Lui, T.; Liu, M.; Erfe, M.C.; Craft, N.; et al. The diversity and host interactions of Propionibacterium acnes bacteriophages on human skin. ISME J. 2015, 9, 2078–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tett, A.; Pasolli, E.; Farina, S.; Truong, D.T.; Asnicar, F.; Zolfo, M.; Beghini, F.; Armanini, F.; Jousson, O.; De Sanctis, V.; et al. Unexplored diversity and strain-level structure of the skin microbiome associated with psoriasis. NPJ Biofilms Microb. 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- McCann, A.; Ryan, F.J.; Stockdale, S.R.; Dalmasso, M.; Blake, T.; Ryan, C.A.; Stanton, C.; Mills, S.; Ross, P.R.; Hill, C. Viromes of one year old infants reveal the impact of birth mode on microbiome diversity. Peer J. 2018, 6, e4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Grunberg, S.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Hypervariable loci in the human gut virome. Proc. Natl. Acad. Sci. USA 2012, 109, 3962–3966. [Google Scholar] [CrossRef] [Green Version]
- Broecker, F.; Russo, G.; Klumpp, J.; Moelling, K. Stable core virome despite variable microbiome after fecal transfer. Gut Microb. 2017, 8, 214–220. [Google Scholar] [CrossRef]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, S.J.; Schreiber, S. Reduced microbial diversity in inflammatory bowel diseases. Gut 2006, 55, 1207. [Google Scholar] [PubMed]
- Deng, Z.L.; Gottschick, C.; Bhuju, S.; Masur, C.; Abels, C.; Wagner-Döbler, I. Metatranscriptome Analysis of the Vaginal Microbiota Reveals Potential Mechanisms for Protection against Metronidazole in Bacterial Vaginosis. mSphere 2018, 3, 1–16. [Google Scholar] [CrossRef]
- Macklaim, J.M.; Fernandes, A.D.; Di Bella, J.M.; Hammond, J.A.; Reid, G.; Gloor, G.B. Comparative meta-RNA-seq of the vaginal microbiota and differential expression by Lactobacillus iners in health and dysbiosis. Microbiome 2013, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gosmann, C.; Anahtar, M.N.; Handley, S.A.; Farcasanu, M.; Abu-Ali, G.; Bowman, B.A.; Padavattan, N.; Desai, C.; Droit, L.; Moodley, A.; et al. Lactobacillus -Deficient Cervicovaginal Bacterial Communities Are Associated with Increased HIV Acquisition in Young South African Women. Immunity 2017, 46, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Rho, M.; Wu, Y.W.; Tang, H.; Doak, T.G.; Ye, Y. Diverse CRISPRs evolving in human microbiomes. PLoS Genet. 2012, 8, e1002441. [Google Scholar] [CrossRef] [PubMed]
- Malki, K.; Shapiro, J.W.; Price, T.K.; Hilt, E.E.; Thomas-White, K.; Sircar, T.; Rosenfeld, A.B.; Kuffel, G.; Zilliox, M.J.; Wolfe, A.J.; et al. Genomes of Gardnerella strains reveal an abundance of prophages within the bladder microbiome. PLoS ONE 2016, 11, 1–16. [Google Scholar] [CrossRef]
- Abdelmaksoud, A.A.; Koparde, V.N.; Sheth, N.U.; Serrano, M.G.; Glascock, A.L.; Fettweis, J.M.; Strauss, J.F.; Buck, G.A.; Jefferson, K.K. Comparison of Lactobacillus crispatus isolates from Lactobacillus-dominated vaginal microbiomes with isolates from microbiomes containing bacterial vaginosis-associated bacteria. Microbiology 2016, 162, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Resource Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.J.; Lynch, D.B.; Murphy, K.; Ulaszewska, M.; Jeffery, I.B.; O’Shea, C.A.; Watkins, C.; Dempsey, E.; Mattivi, F.; Tuohy, K.; et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome 2017, 5, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alipour, M.; Zaidi, D.; Valcheva, R.; Jovel, J.; Martínez, I.; Sergi, C.; Walter, J.; Mason, A.L.; Wong, G.K.; Dieleman, L.A.; et al. Mucosal barrier depletion and loss of bacterial diversity are primary abnormalities in paediatric ulcerative colitis. J. Crohns Colitis 2016, 10, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2016, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Kleiner, M.; Paez-Espino, D.; Zhu, W.; Bushnell, B.; Hassell, B.; Winter, S.E.; Kyrpides, N.C.; Hooper, L.V. Murine colitis reveals a disease-associated bacteriophage community. Nat. Microbiol. 2018, 3, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microb. 2019, 25, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Baker, K.; Padman, B.S.; Patwa, R.; Dunstan, R.A.; Weston, T.A.; Schlosser, K.; Bailey, B.; Lithgow, T.; Lazarou, M.; et al. Bacteriophage Transcytosis Provides a Mechanism To Cross Epithelial Cell Layers. MBio. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile Colitis. N. Engl. J. Med. 1994, 330, 257–262. [Google Scholar] [CrossRef]
- Fortier, L.C. Bacteriophages contribute to shaping clostridioides (Clostridium) difficile species. Front. Microbiol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Amy, J.; Bulach, D.; Knight, D.; Riley, T.; Johanesen, P.; Lyras, D. Identification of large cryptic plasmids in Clostridioides (Clostridium) difficile. Plasmid 2018, 96–97, 25–38. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2015, 14, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Hannigan, G.D.; Meisel, J.S.; Tyldsley, A.S.; Zheng, Q.; Hodkinson, B.P.; SanMiguel, A.J.; Minot, S.; Bushman, F.D.; Grice, E.A. The Human Skin Double-Stranded DNA Virome: Topographical and Temporal Diversity, Genetic Enrichment, and Dynamic Associations with the Host Microbiome. mBio 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Van de Wijgert, J.H.; Borgdorff, H.; Verhelst, R.; Crucitti, T.; Francis, S.; Verstraelen, H.; Jespers, V. The vaginal microbiota: What have we learned after a decade of molecular characterization? PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Jerne, N.K. Bacteriophage inactivation by antiphage serum diluted in distilled water. Nature 1952, 169, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Kaźmierczak, Z.; Letarov, A.; Górski, A. Immunogenicity studies of proteins forming the T4 phage head surface. J. Virol. 2014, 88, 12551–12557. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Smith, M.C.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawrence, D.; Baldridge, M.T.; Handley, S.A. Phages and Human Health: More Than Idle Hitchhikers. Viruses 2019, 11, 587. https://doi.org/10.3390/v11070587
Lawrence D, Baldridge MT, Handley SA. Phages and Human Health: More Than Idle Hitchhikers. Viruses. 2019; 11(7):587. https://doi.org/10.3390/v11070587
Chicago/Turabian StyleLawrence, Dylan, Megan T. Baldridge, and Scott A. Handley. 2019. "Phages and Human Health: More Than Idle Hitchhikers" Viruses 11, no. 7: 587. https://doi.org/10.3390/v11070587
APA StyleLawrence, D., Baldridge, M. T., & Handley, S. A. (2019). Phages and Human Health: More Than Idle Hitchhikers. Viruses, 11(7), 587. https://doi.org/10.3390/v11070587