Phylogeography of Puumala orthohantavirus in Europe

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Acquisition
2.2. Phylogenetic Analyses
2.3. Phylogeographic Analyses
2.3.1. Bayesian Method
2.3.2. Maximum Likelihood Methods
2.4. Visualization and Analyses of the PUUV Dispersion Pathways
3. Results
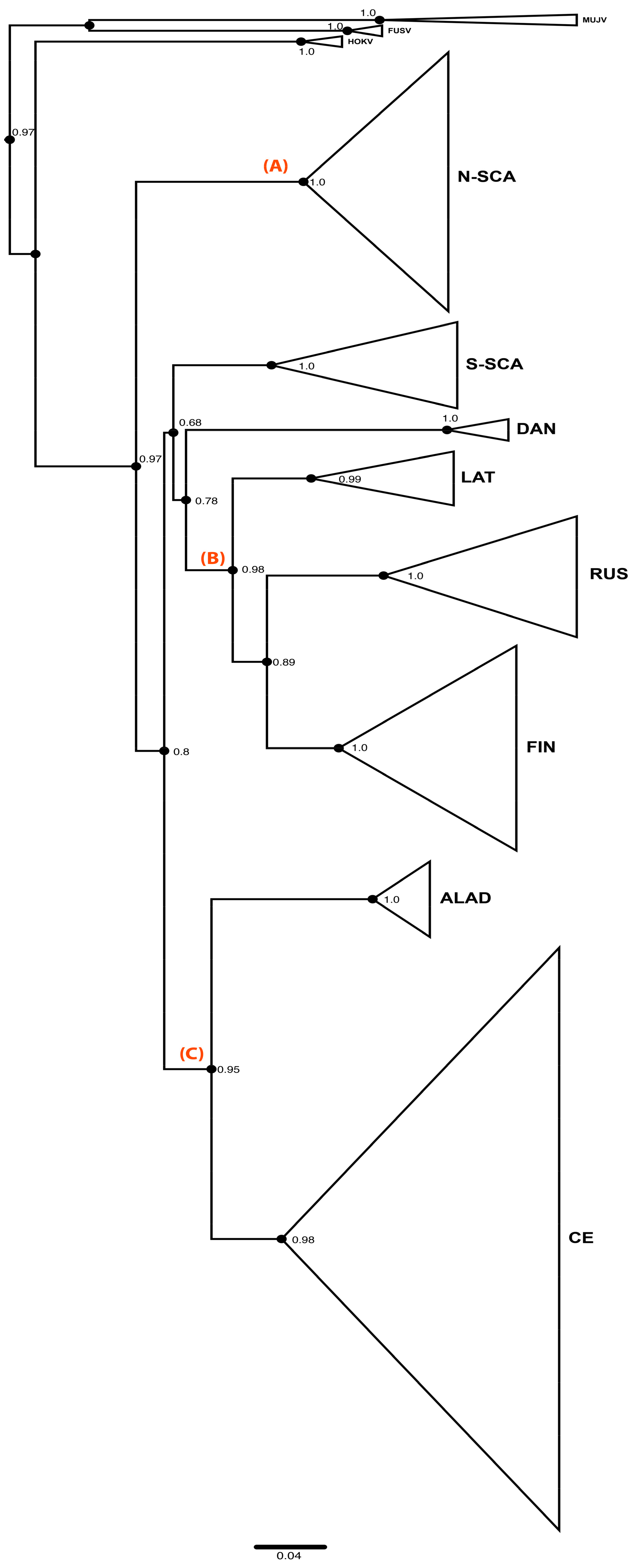
3.1. Phylogenetic Analyses of PUUV S Segment Sequence Dataset
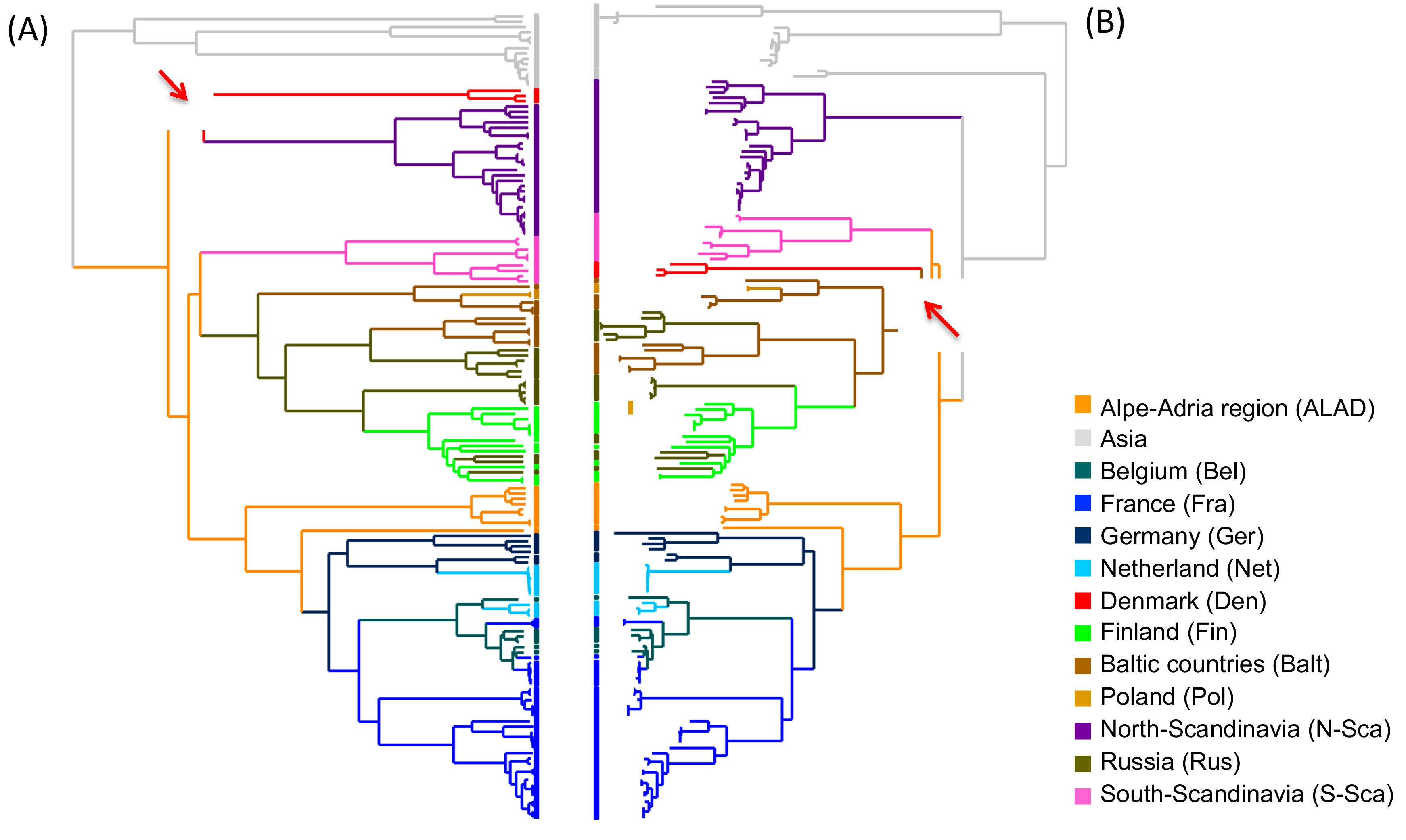
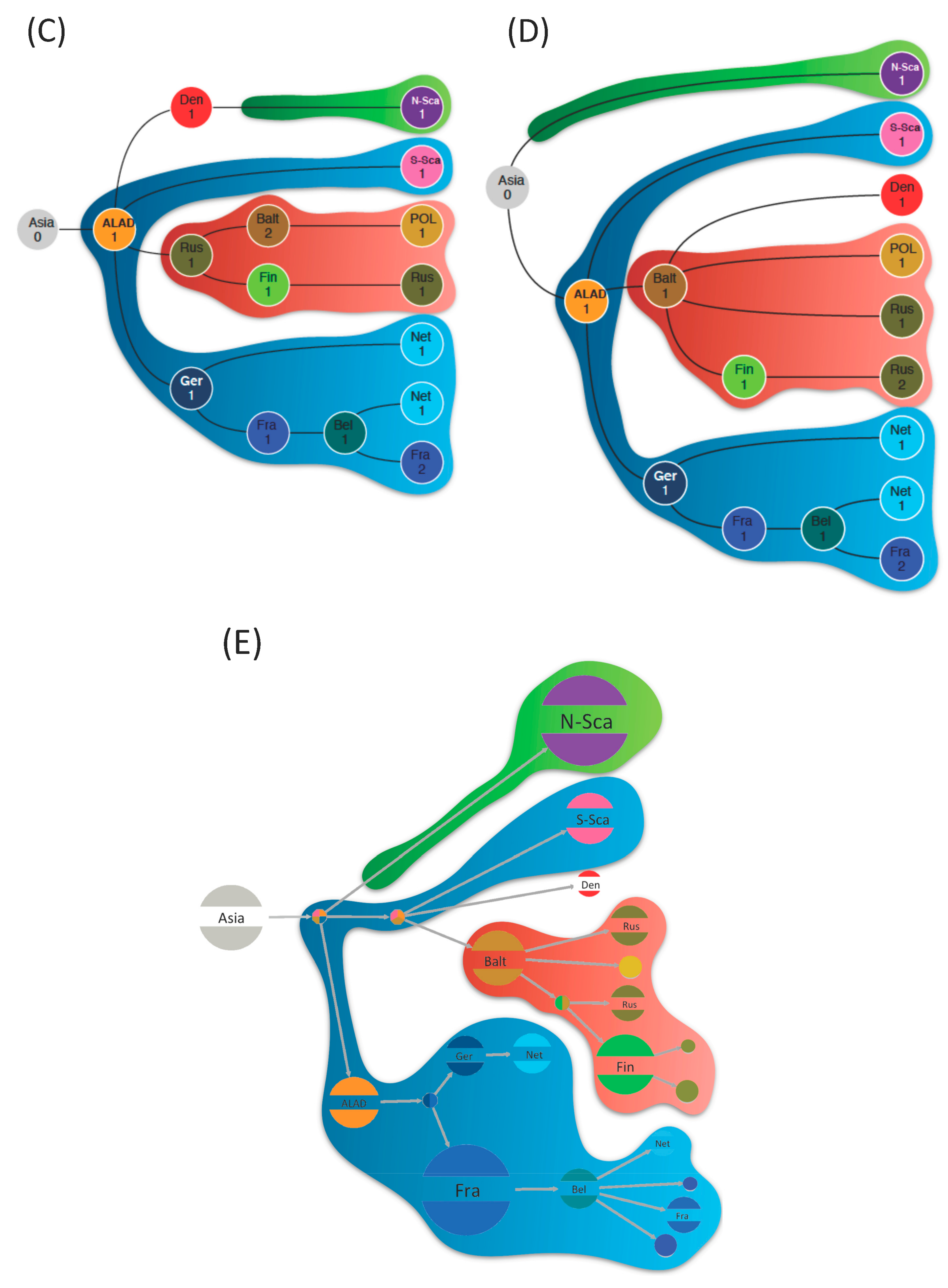
3.2. Phylogeographic Reconstructions
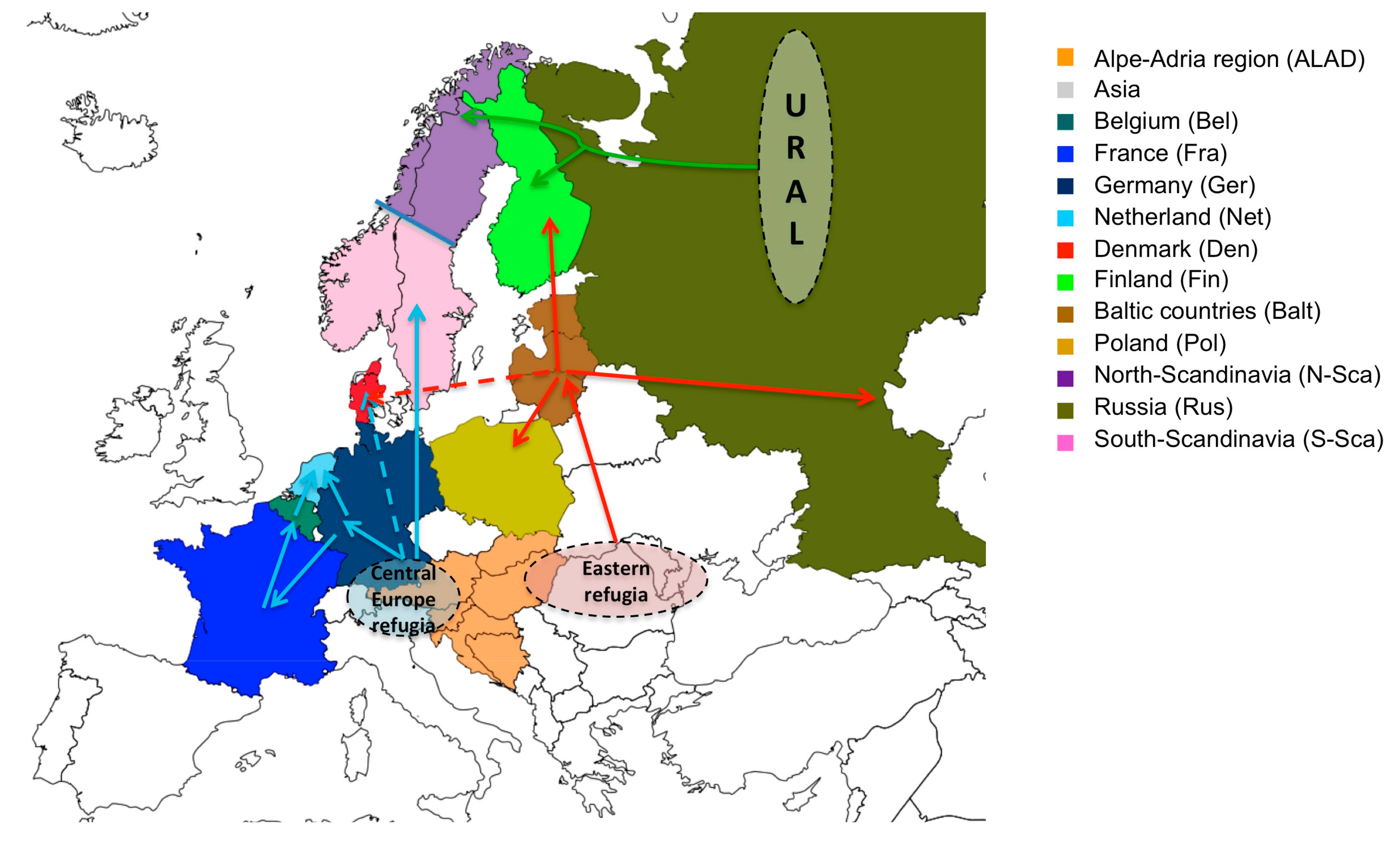
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Maes, P.; Adkins, S.; Alkhovsky, S.V.; Avsic-Zupanc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, E.; Blair, C.D.; Briese, T.; et al. Taxonomy of the order Bunyavirales: Second update 2018. Arch. Virol. 2019, 164, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Brummer-Korvenkontio, M.; Vaheri, A.; Hovi, T.; von Bonsdorff, C.H.; Vuorimies, J.; Manni, T.; Penttinen, K.; Oker-Blom, N.; Lahdevirta, J. Nephropathia epidemica: Detection of antigen in bank voles and serologic diagnosis of human infection. J. Infect. Dis. 1980, 141, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Lee, P.W.; Johnson, K.M. Isolation of the etiologic agent of Korean Hemorrhagic fever. J. Infect. Dis. 1978, 137, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, A. Genetics of hantaviruses: Implications to taxonomy. Arch. Virol. 2002, 147, 665–682. [Google Scholar] [CrossRef] [PubMed]
- Jaaskelainen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus infections in Europe and their impact on public health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, R.; Krystufek, B.; Yigit, N.; Mitsain, G.; Palomo, L.J.; Henttonen, H.; Vohralik, V.; Zagorodnyuk, I.; Juskaitis, R.; Meinig, H.; et al. Myodes glareolus (Errata Version Published in 2017). The IUCN Red List of Threatened Species 2016. Available online: http://dx.doi.org/10.2305/IUCN.UK.2016-3.RLTS.T4973A22372716.en (accessed on 01 May 2019).
- Kallio, E.R.; Begon, M.; Henttonen, H.; Koskela, E.; Mappes, T.; Vaheri, A.; Vapalahti, O. Cyclic hantavirus epidemics in humans--predicted by rodent host dynamics. Epidemics 2009, 1, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Makary, P.; Kanerva, M.; Ollgren, J.; Virtanen, M.J.; Vapalahti, O.; Lyytikainen, O. Disease burden of Puumala virus infections, 1995–2008. Epidemiol. Infect. 2010, 138, 1484–1492. [Google Scholar] [CrossRef]
- Reynes, J.M.; Dutrop, C.M.; Carli, D.; Levast, M.; Fontaine, N.; Denoyel, G.A.; Philit, J.B. Puumala hantavirus infection in Isere: Geographic extension of this zoonosis in France. Med. Mal. Infect. 2015, 45, 177–180. [Google Scholar] [CrossRef]
- Olsson, G.E.; Leirs, H.; Henttonen, H. Hantaviruses and their hosts in Europe: Reservoirs here and there, but not everywhere? Vector Borne Zoonotic Dis. 2010, 10, 549–561. [Google Scholar] [CrossRef]
- Heyman, P.; Vaheri, A.; Lundkvist, A.; Avsic-Zupanc, T. Hantavirus infections in Europe: From virus carriers to a major public-health problem. Expert Rev. Anti-Infect. Ther. 2009, 7, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Sironen, T.; Vaheri, A.; Plyusnin, A. Molecular evolution of Puumala hantavirus. J. Virol. 2001, 75, 11803–11810. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Niemimaa, J.; Henttonen, H.; Plyusnin, A. Co-circulation of two Puumala hantavirus lineages in Latvia: A Russian lineage described previously and a novel Latvian lineage. J. Med. Virol. 2012, 84, 314–318. [Google Scholar] [PubMed]
- Plyusnin, A.; Vapalahti, O.; Ulfves, K.; Lehvaslaiho, H.; Apekina, N.; Gavrilovskaya, I.; Blinov, V.; Vaheri, A. Sequences of wild Puumala virus genes show a correlation of genetic variation with geographic origin of the strains. J. Gen. Virol. 1994, 75 Pt 2, 405–409. [Google Scholar] [CrossRef]
- Plyusnina, A.; Razzauti, M.; Sironen, T.; Niemimaa, J.; Vapalahti, O.; Vaheri, A.; Henttonen, H.; Plyusnin, A. Analysis of complete Puumala virus genome, Finland. Emerg. Infect. Dis. 2012, 18, 2070–2072. [Google Scholar] [CrossRef] [PubMed]
- Asikainen, K.; Hanninen, T.; Henttonen, H.; Niemimaa, J.; Laakkonen, J.; Andersen, H.K.; Bille, N.; Leirs, H.; Vaheri, A.; Plyusnin, A. Molecular evolution of Puumala hantavirus in Fennoscandia: Phylogenetic analysis of strains from two recolonization routes, Karelia and Denmark. J. Gen. Virol. 2000, 81, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, A.; Morzunov, S.P. Virus evolution and genetic diversity of hantaviruses and their rodent hosts. Curr. Top. Microbiol. Immunol. 2001, 256, 47–75. [Google Scholar] [PubMed]
- Plyusnin, A.; Sironen, T. Evolution of hantaviruses: Co-speciation with reservoir hosts for more than 100 MYR. Virus Res. 2014, 187, 22–26. [Google Scholar] [PubMed]
- Jackson, A.P.; Charleston, M.A. A cophylogenetic perspective of RNA-virus evolution. Mol. Biol. Evol. 2004, 21, 45–57. [Google Scholar] [PubMed]
- Castel, G.; Tordo, N.; Plyusnin, A. Estimation of main diversification time-points of hantaviruses using phylogenetic analyses of complete genomes. Virus Res. 2017, 233, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.; Holmes, E.C.; Charleston, M.A. Hantavirus evolution in relation to its rodent and insectivore hosts: No evidence for codivergence. Mol. Biol. Evol. 2009, 26, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.; Melo, F.L.; Figueiredo, L.M.; Holmes, E.C.; Zanotto, P.M. High rates of molecular evolution in hantaviruses. Mol. Biol. Evol. 2008, 25, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, A.; Vapalahti, O.; Lehvaslaiho, H.; Apekina, N.; Mikhailova, T.; Gavrilovskaya, I.; Laakkonen, J.; Niemimaa, J.; Henttonen, H.; Brummer-Korvenkontio, M.; et al. Genetic variation of wild Puumala viruses within the serotype, local rodent populations and individual animal. Virus Res. 1995, 38, 25–41. [Google Scholar] [CrossRef]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Accumulation of point mutations and reassortment of genomic RNA segments are involved in the microevolution of Puumala hantavirus in a bank vole (Myodes glareolus) population. J. Gen. Virol. 2008, 89, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Microevolution of Puumala hantavirus during a complete population cycle of its host, the bank vole (Myodes glareolus). PLoS ONE 2013, 8, e64447. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Sironen, T.; Henttonen, H.; Plyusnin, A. Analysis of Puumala hantavirus in a bank vole population in northern Finland: Evidence for co-circulation of two genetic lineages and frequent reassortment between strains. J. Gen. Virol. 2009, 90, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Klempa, B. Reassortment events in the evolution of hantaviruses. Virus Genes 2018, 54, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chare, E.R.; Gould, E.A.; Holmes, E.C. Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 2003, 84, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Friedman, R. Evolutionary diversification of protein-coding genes of hantaviruses. Mol. Biol. Evol. 2000, 17, 1558–1568. [Google Scholar] [CrossRef]
- Castel, G.; Razzauti, M.; Jousselin, E.; Kergoat, G.J.; Cosson, J.F. Changes in diversification patterns and signatures of selection during the evolution of murinae-associated hantaviruses. Viruses 2014, 6, 1112–1134. [Google Scholar] [CrossRef]
- Laenen, L.; Vergote, V.; Vanmechelen, B.; Tersago, K.; Baele, G.; Lemey, P.; Leirs, H.; Dellicour, S.; Vrancken, B.; Maes, P. Identifying the patterns and drivers of Puumala hantavirus enzootic dynamics using reservoir sampling. Virus Evol. 2019, 5, vez009. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.N.; Gu, S.H.; Kang, H.J.; Arai, S.; Yanagihara, R. Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 2014, 22, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxenhofer, M.; Weber de Melo, V.; Ulrich, R.G.; Heckel, G. Revised time scales of RNA virus evolution based on spatial information. Proc. R. Soc. B Biol. Sci. 2017, 284, 20170857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, W.M.; Bello, G.; Amarilla, A.A.; Alfonso, H.L.; Aquino, V.H.; Figueiredo, L.T. Phylogeography and evolutionary history of rodent-borne hantaviruses. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2014, 21, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Zhang, Y.Z. The evolution and emergence of hantaviruses. Curr. Opin. Virol. 2015, 10, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Suchard, M.A.; Rambaut, A.; Lemey, P. Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 2011, 1, 423–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monchatre-Leroy, E.; Murri, S.; Castel, G.; Calavas, D.; Boue, F.; Henaux, V.; Marianneau, P. First insights into Puumala orthohantavirus circulation in a rodent population in Alsace, France. Zoonoses Public Health 2018, 65, 540–551. [Google Scholar] [CrossRef]
- Lee, J.G.; Gu, S.H.; Baek, L.J.; Shin, O.S.; Park, K.S.; Kim, H.C.; Klein, T.A.; Yanagihara, R.; Song, J.W. Muju virus, harbored by Myodes regulus in Korea, might represent a genetic variant of Puumala virus, the prototype arvicolid rodent-borne hantavirus. Viruses 2014, 6, 1701–1714. [Google Scholar] [CrossRef]
- Plyusnina, A.; Laakkonen, J.; Niemimaa, J.; Nemirov, K.; Muruyeva, G.; Pohodiev, B.; Lundkvist, A.; Vaheri, A.; Henttonen, H.; Vapalahti, O.; et al. Genetic analysis of hantaviruses carried by Myodes and Microtus rodents in Buryatia. Virol. J. 2008, 5, 4. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Zou, Y.; Yan, Y.Z.; Hu, G.W.; Yao, L.S.; Du, Z.S.; Jin, L.Z.; Liu, Y.Y.; Li, M.H.; Chen, H.X.; et al. Detection of phylogenetically distinct Puumala-like viruses from red-grey vole Clethrionomys rufocanus in China. J. Med. Virol. 2007, 79, 1208–1218. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SMS: Smart Model Selection in PhyML. Available online: http://www.atgc-montpellier.fr/sms/ (accessed on 01 April 2019).
- Rambaut, A. FigTree, 1.4. 2009. Available online: tree.bio.ed.ac.uk/software/figtree/ (accessed date 29 May 2019).
- Gascuel, O.; Steel, M. Predicting the ancestral character changes in a tree is typically easier than predicting the root state. Syst. Biol. 2014, 63, 421–435. [Google Scholar] [CrossRef]
- Royer-Carenzi, M.; Pontarotti, P.; Didier, G. Choosing the best ancestral character state reconstruction method. Math. Biosci. 2013, 242, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, A.M.; Lo, N.; Ho, S.Y.W. The Impact of the Tree Prior on Molecular Dating of Data Sets Containing a Mixture of Inter- and Intraspecies Sampling. Syst. Biol. 2017, 66, 413–425. [Google Scholar] [CrossRef]
- Ho, S.Y.; Shapiro, B. Skyline-plot methods for estimating demographic history from nucleotide sequences. Mol. Ecol. Resour. 2011, 11, 423–434. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Ferreira, M.A.R.; Suchard, M. Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 2008, 36, 355–368. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevenet, F.; Castel, G.; Jousselin, E.; Gascuel, O. PastView: A user-friendly interface to explore evolutionary scenarios. BioRxiv 2019. [Google Scholar]
- PastView. Available online: http://web-mab.lirmm.fr/pastview/ (accessed on 01 April 2019).
- PastML. Available online: https://pastml.pasteur.fr/ (accessed on 01 April 2019).
- Ishikawa, S.A.; Zhukova, A.; Iwasaki, W.; Gascuel, O. A Fast Likelihood Method to Reconstruct and Visualize Ancestral Scenarios. Mol. Biol. Evol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Cazaux, B.; Castel, G.; Rivals, E. AQUAPONY: Visualization and interpretation of phylogeographic information on phylogenetic trees. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- AQUAPONY. Available online: http://www.atgc-montpellier.fr/aquapony/ (accessed on 01 April 2019).
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plyusnina, A.; Deter, J.; Charbonnel, N.; Cosson, J.F.; Plyusnin, A. Puumala and Tula hantaviruses in France. Virus Res. 2007, 129, 58–63. [Google Scholar] [CrossRef]
- Castel, G.; Couteaudier, M.; Sauvage, F.; Pons, J.B.; Murri, S.; Plyusnina, A.; Pontier, D.; Cosson, J.F.; Plyusnin, A.; Marianneau, P.; et al. Complete Genome and Phylogeny of Puumala hantavirus Isolates Circulating in France. Viruses 2015, 7, 5476–5488. [Google Scholar] [CrossRef]
- Escutenaire, S.; Chalon, P.; Heyman, P.; Van der Auwera, G.; van der Groen, G.; Verhagen, R.; Thomas, I.; Karelle-Bui, L.; Vaheri, A.; Pastoret, P.P.; et al. Genetic characterization of Puumala hantavirus strains from Belgium: Evidence for a distinct phylogenetic lineage. Virus Res. 2001, 74, 1–15. [Google Scholar] [CrossRef]
- Sharp, P.M.; Simmonds, P. Evaluating the evidence for virus/host co-evolution. Curr. Opin. Virol. 2011, 1, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Laenen, L.; Dellicour, S.; Vergote, V.; Nauwelaers, I.; De Coster, S.; Verbeeck, I.; Vanmechelen, B.; Lemey, P.; Maes, P. Spatio-temporal analysis of Nova virus, a divergent hantavirus circulating in the European mole in Belgium. Mol. Ecol. 2016, 25, 5994–6008. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Kosakovsky Pond, S.L. Purifying selection can obscure the ancient age of viral lineages. Mol. Biol. Evol. 2011, 28, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.J.; Ballinger, M.J.; Zhan, J.J.; Hanzly, L.E.; Bruenn, J.A. Evidence that ebolaviruses and cuevaviruses have been diverging from marburgviruses since the Miocene. PeerJ 2014, 2, e556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, E.C. Evolutionary history and phylogeography of human viruses. Annu. Rev. Microbiol. 2008, 62, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Holmes, E.C. What is the time-scale of hantavirus evolution? Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2014, 25, 144–145. [Google Scholar] [CrossRef] [PubMed]
- Biek, R.; Drummond, A.J.; Poss, M. A virus reveals population structure and recent demographic history of its carnivore host. Science 2006, 311, 538–541. [Google Scholar] [CrossRef]
- Pybus, O.G.; Rambaut, A. Evolutionary analysis of the dynamics of viral infectious disease. Nat. Rev. Genet. 2009, 10, 540–550. [Google Scholar] [CrossRef]
- Nieberding, C.M.; Durette-Desset, M.C.; Vanderpoorten, A.; Casanova, J.C.; Ribas, A.; Deffontaine, V.; Feliu, C.; Morand, S.; Libois, R.; Michaux, J.R. Geography and host biogeography matter for understanding the phylogeography of a parasite. Mol. Phylogenet. Evol. 2008, 47, 538–554. [Google Scholar] [CrossRef]
- Nemirov, K.; Leirs, H.; Lundkvist, A.; Olsson, G.E. Puumala hantavirus and Myodes glareolus in northern Europe: No evidence of co-divergence between genetic lineages of virus and host. J. Gen. Virol. 2010, 91, 1262–1274. [Google Scholar] [CrossRef]
- Runck, A.M.; Cook, J.A. Postglacial expansion of the southern red-backed vole (Clethrionomys gapperi) in North America. Mol. Ecol. 2005, 14, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Ledevin, R.; Quere, J.P.; Renaud, S. Morphometrics as an insight into processes beyond tooth shape variation in a bank vole population. PLoS ONE 2010, 5, e15470. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.G.; Cosson, J.F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Jaarola, M.; Searle, J.B. Phylogeography of field voles (Microtus agrestis) in Eurasia inferred from mitochondrial DNA sequences. Mol. Ecol. 2002, 11, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Kotlik, P.; Deffontaine, V.; Mascheretti, S.; Zima, J.; Michaux, J.R.; Searle, J.B. A northern glacial refugium for bank voles (Clethrionomys glareolus). Proc. Natl. Acad. Sci. USA 2006, 103, 14860–14864. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, J.M.; Kawalko, A.; Markova, S.; Searle, J.B.; Kotlik, P. Phylogeographic signatures of northward post-glacial colonization from high-latitude refugia: A case study of bank voles using museum specimens. J. Zool. 2010, 281, 249–262. [Google Scholar] [CrossRef]
- Deffontaine, V.; Libois, R.; Kotlik, P.; Sommer, R.; Nieberding, C.; Paradis, E.; Searle, J.B.; Michaux, J.R. Beyond the Mediterranean peninsulas: Evidence of central European glacial refugia for a temperate forest mammal species, the bank vole (Clethrionomys glareolus). Mol. Ecol. 2005, 14, 1727–1739. [Google Scholar] [CrossRef]
- Deffontaine, V.; Ledevin, R.; Fontaine, M.C.; Quere, J.P.; Renaud, S.; Libois, R.; Michaux, J.R. A relict bank vole lineage highlights the biogeographic history of the Pyrenean region in Europe. Mol. Ecol. 2009, 18, 2489–2502. [Google Scholar] [CrossRef] [Green Version]
- Bilton, D.T.; Mirol, P.M.; Mascheretti, S.; Fredga, K.; Zima, J.; Searle, J.B. Mediterranean Europe as an area of endemism for small mammals rather than a source for northwards postglacial colonization. Proc. Biol. Sci. 1998, 265, 1219–1226. [Google Scholar] [CrossRef]
- Jaarola, M.; Tegelstrom, H. Colonization history of north European field voles (Microtus agrestis) revealed by mitochondrial DNA. Mol. Ecol. 1995, 4, 299–310. [Google Scholar] [CrossRef]
- Tarnowska, E.; Niedzialkowska, M.; Gerc, J.; Korbut, Z.; Gorny, M.; Jedrzejewska, B. Spatial distribution of the Carpathian and Eastern mtDNA lineages of the bank vole in their contact zone relates to environmental conditions. Biol. J. Linn. Soc. 2016, 119, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Abramson, N.I.; Rodchenkova, E.N.; Kostygov, A. Genetic variation and phylogeography of the bank vole (Clethrionomys glareolus, Arvicolinae, Rodentia) in Russia with special reference to the introgression of the mtDNA of a closely related species, red-backed vole (C. rutilus). Genetika 2009, 45, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Tegelstrom, H. Transfer of mitochondrial DNA from the northern red-backed vole (Clethrionomys rutilus) to the bank vole (C. glareolus). J. Mol. Evol. 1987, 24, 218–227. [Google Scholar] [CrossRef]
- Jaarola, M.; Tegelstrom, H.; Fredga, K. Colonization history in Fennoscandian rodents. Biol. J. Linn. Soc. 1999, 68, 113–127. [Google Scholar] [CrossRef]
- Boratynski, Z.; Alves, P.C.; Berto, S.; Koskela, E.; Mappes, T.; Melo-Ferreira, J. Introgression of mitochondrial DNA among Myodes voles: Consequences for energetics? BMC Evol. Biol. 2011, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Strakova, P.; Jagdmann, S.; Balciauskas, L.; Balciauskiene, L.; Drewes, S.; Ulrich, R.G. Puumala Virus in Bank Voles, Lithuania. Emerg. Infect. Dis. 2017, 23, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M. Microevolution of Puumala hantavirus in its host, the bank vole (Myodes glareolus). Ph.D. Thesis, University of Helsinki, Helsinki, Finland, 2012. [Google Scholar]
- Horling, J.; Lundkvist, A.; Jaarola, M.; Plyusnin, A.; Tegelstrom, H.; Persson, K.; Lehvaslaiho, H.; Hornfeldt, B.; Vaheri, A.; Niklasson, B. Distribution and genetic heterogeneity of Puumala virus in Sweden. J. Gen. Virol. 1996, 77 Pt 10, 2555–2562. [Google Scholar] [CrossRef]
- Dekonenko, A.; Yakimenko, V.; Ivanov, A.; Morozov, V.; Nikitin, P.; Khasanova, S.; Dzagurova, T.; Tkachenko, E.; Schmaljohn, C. Genetic similarity of Puumala viruses found in Finland and western Siberia and of the mitochondrial DNA of their rodent hosts suggests a common evolutionary origin. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2003, 3, 245–257. [Google Scholar] [CrossRef]
- Bernshtein, A.D.; Apekina, N.S.; Mikhailova, T.V.; Myasnikov, Y.A.; Khlyap, L.A.; Korotkov, Y.S.; Gavrilovskaya, I.N. Dynamics of Puumala hantavirus infection in naturally infected bank voles (Clethrinomys glareolus). Arch. Virol. 1999, 144, 2415–2428. [Google Scholar] [CrossRef]
- Sehrawat, S.; Kumar, D.; Rouse, B.T. Herpesviruses: Harmonious Pathogens but Relevant Cofactors in Other Diseases? Front. Cell. Infect. Microbiol. 2018, 8, 177. [Google Scholar] [CrossRef]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Dubois, A.; Castel, G.; Murri, S.; Pulido, C.; Pons, J.B.; Benoit, L.; Loiseau, A.; Lakhdar, L.; Galan, M.; Charbonnel, N.; et al. Experimental infections of wild bank voles (Myodes glareolus) from nephropatia epidemica endemic and non-endemic regions revealed slight differences in Puumala virological course and immunological responses. Virus Res. 2017, 235, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Kallio, E.R.; Klingstrom, J.; Gustafsson, E.; Manni, T.; Vaheri, A.; Henttonen, H.; Vapalahti, O.; Lundkvist, A. Prolonged survival of Puumala hantavirus outside the host: Evidence for indirect transmission via the environment. J. Gen. Virol. 2006, 87, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Kallio, E.R.; Begon, M.; Henttonen, H.; Koskela, E.; Mappes, T.; Vaheri, A.; Vapalahti, O. Hantavirus infections in fluctuating host populations: The role of maternal antibodies. Proc. Biol. Sci. 2010, 277, 3783–3791. [Google Scholar] [CrossRef] [PubMed]
- Deter, J.; Bryja, J.; Chaval, Y.; Galan, M.; Henttonen, H.; Laakkonen, J.; Voutilainen, L.; Vapalahti, O.; Vaheri, A.; Salvador, A.R.; et al. Association between the DQA MHC class II gene and Puumala virus infection in Myodes glareolus, the bank vole. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2008, 8, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Tersago, K.; Schreurs, A.; Linard, C.; Verhagen, R.; Van Dongen, S.; Leirs, H. Population, environmental, and community effects on local bank vole (Myodes glareolus) Puumala virus infection in an area with low human incidence. Vector Borne Zoonotic Dis. 2008, 8, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Smura, T.; Tamarit, D.; Huitu, O.; Voutilainen, L.; Henttonen, H.; Vaheri, A.; Vapalahti, O.; Sironen, T. Evolution and postglacial colonization of Seewis hantavirus with Sorex araneus in Finland. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2018, 57, 88–97. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castel, G.; Chevenet, F.; Razzauti, M.; Murri, S.; Marianneau, P.; Cosson, J.-F.; Tordo, N.; Plyusnin, A. Phylogeography of Puumala orthohantavirus in Europe. Viruses 2019, 11, 679. https://doi.org/10.3390/v11080679
Castel G, Chevenet F, Razzauti M, Murri S, Marianneau P, Cosson J-F, Tordo N, Plyusnin A. Phylogeography of Puumala orthohantavirus in Europe. Viruses. 2019; 11(8):679. https://doi.org/10.3390/v11080679
Chicago/Turabian StyleCastel, Guillaume, François Chevenet, Maria Razzauti, Séverine Murri, Philippe Marianneau, Jean-François Cosson, Noël Tordo, and Alexander Plyusnin. 2019. "Phylogeography of Puumala orthohantavirus in Europe" Viruses 11, no. 8: 679. https://doi.org/10.3390/v11080679
APA StyleCastel, G., Chevenet, F., Razzauti, M., Murri, S., Marianneau, P., Cosson, J.-F., Tordo, N., & Plyusnin, A. (2019). Phylogeography of Puumala orthohantavirus in Europe. Viruses, 11(8), 679. https://doi.org/10.3390/v11080679