
Application of Next Generation Sequencing (NGS) in Phage Displayed Peptide Selection to Support the Identification of Arsenic-Binding Motifs

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Media and Buffer
2.2. Phage Library
2.3. Biopanning
2.3.1. Experimental Setup
2.3.2. Column Handling and Target Immobilization
2.3.3. Phage Library Application and Enrichment
2.4. Sanger Sequencing
2.5. Illumina Sequencing
2.6. Bioinformatic Processing
2.6.1. Analysis of Illumina Data
2.6.2. Sequence Evaluation
3. Results
3.1. Biopanning Experiments
3.2. Illumina (NGS) Sequencing
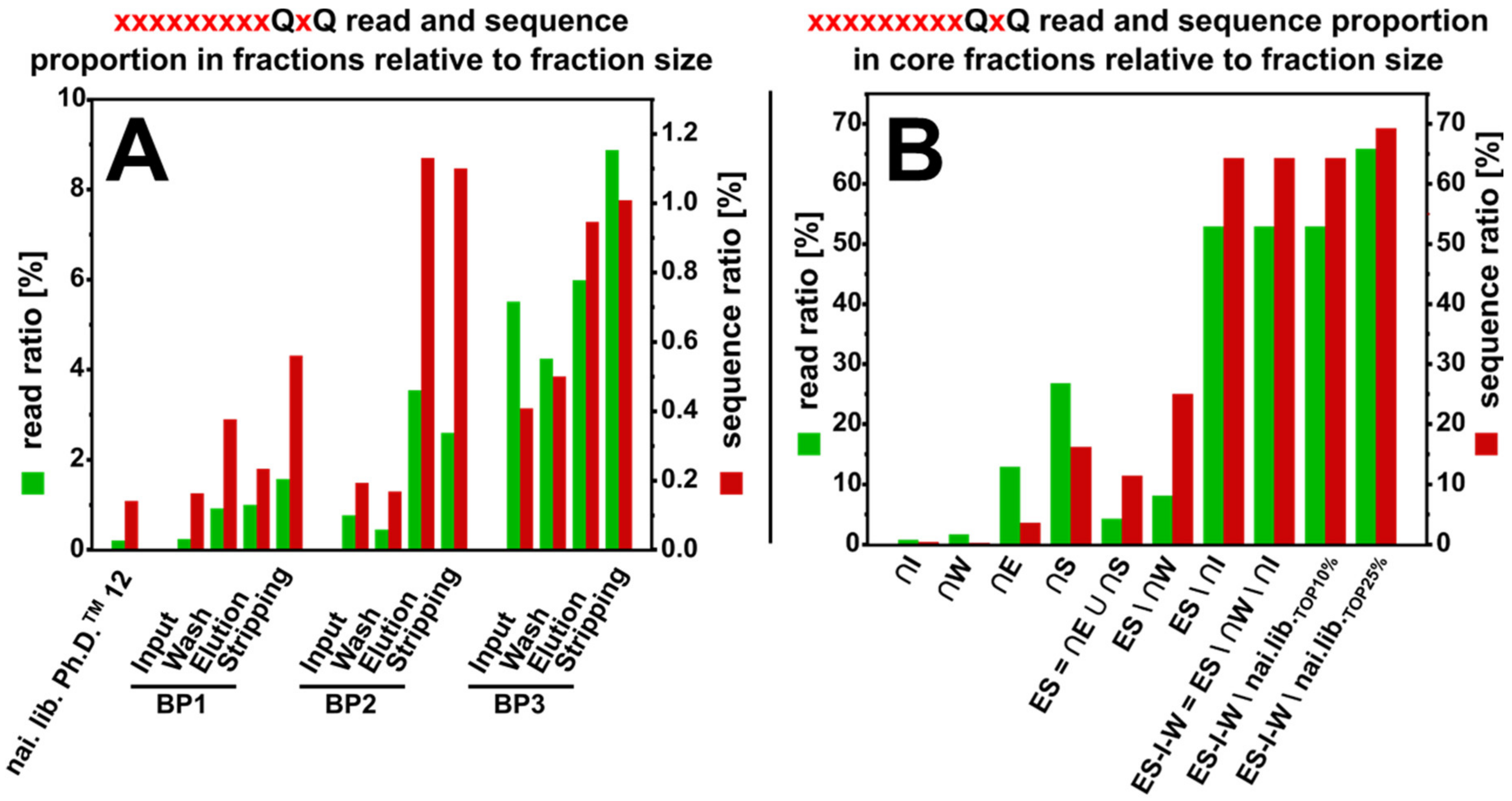
3.3. NGS Fraction Composition
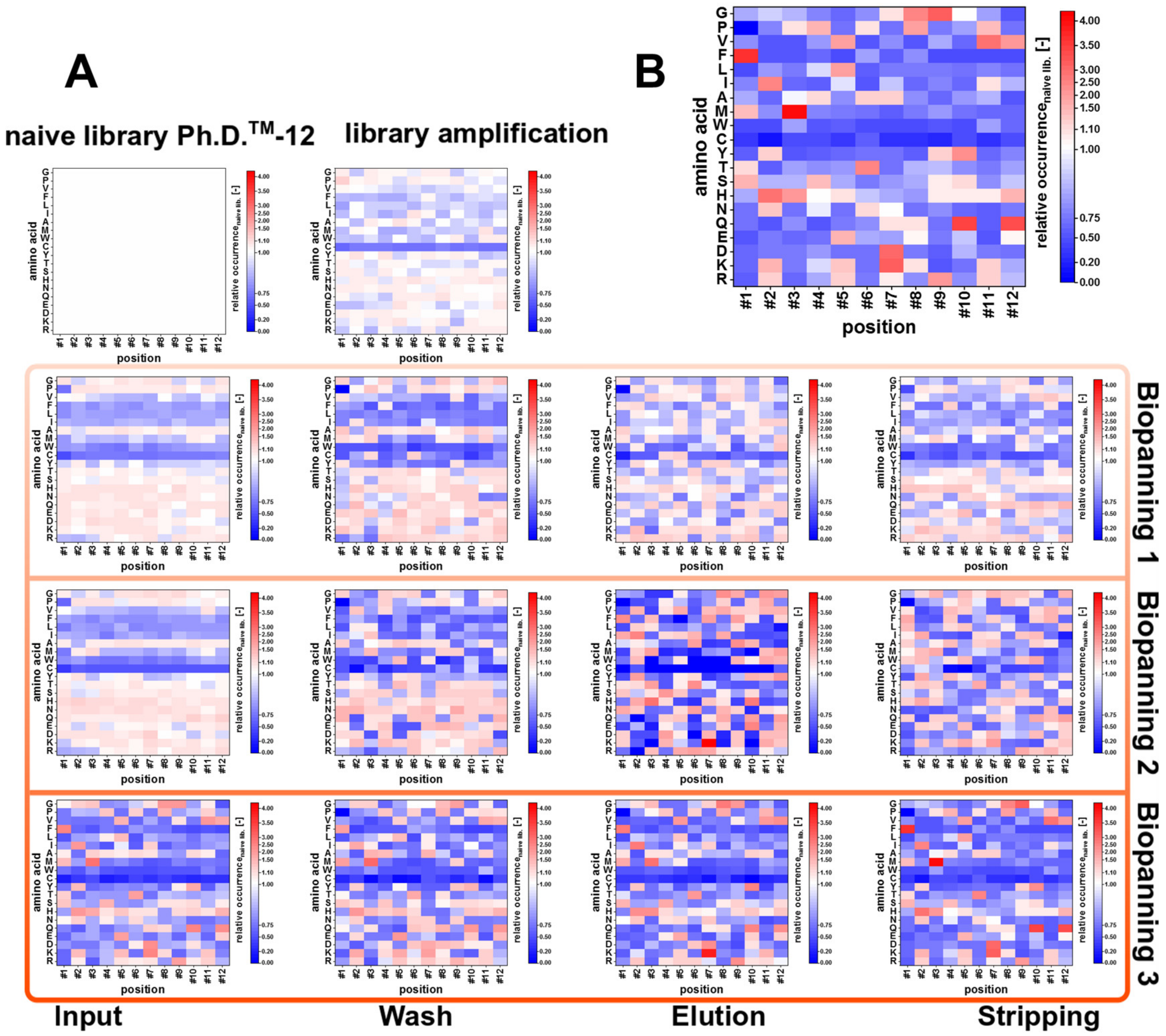
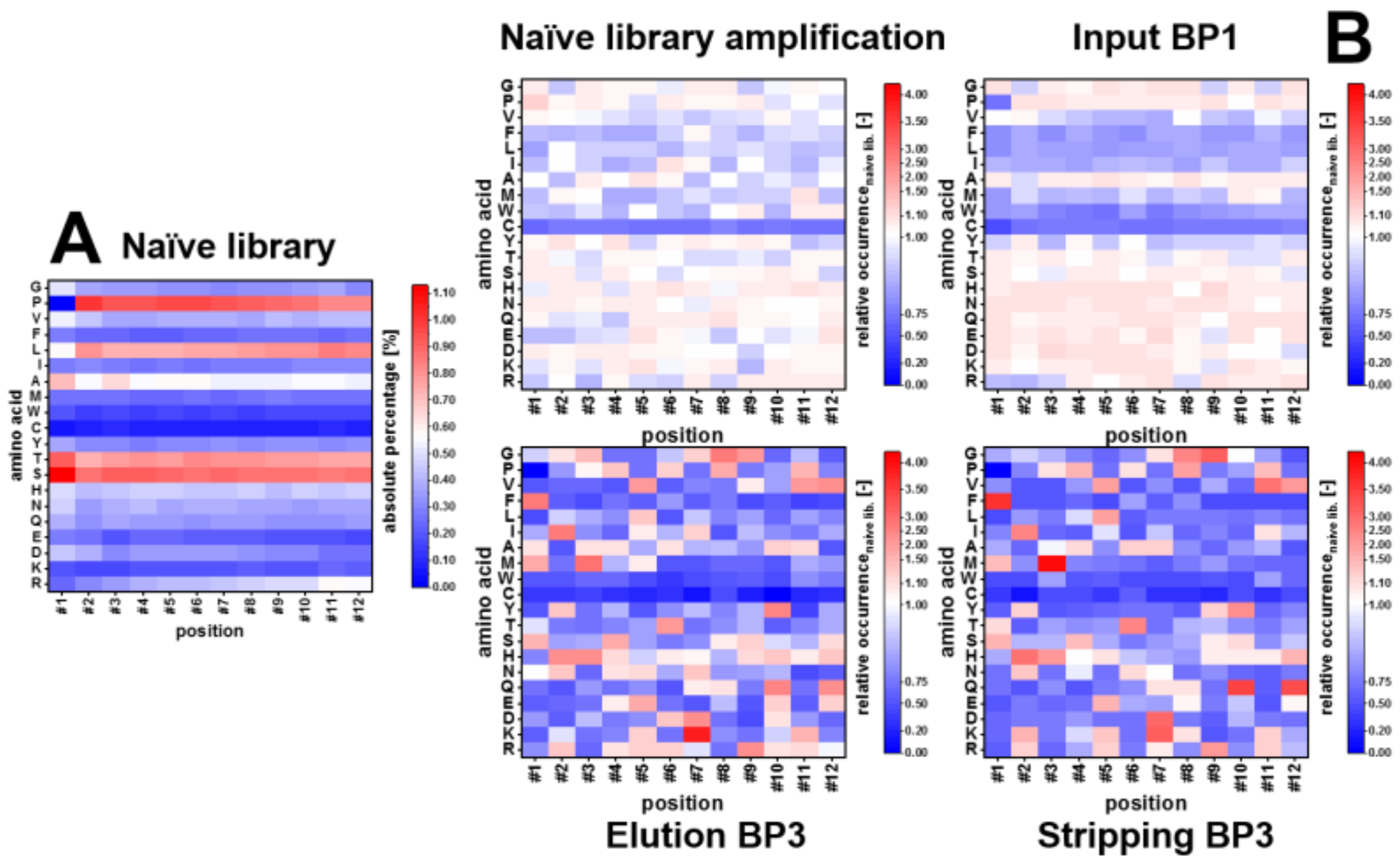
3.4. Amino Acid Composition
3.5. Sequence Logo Calculation
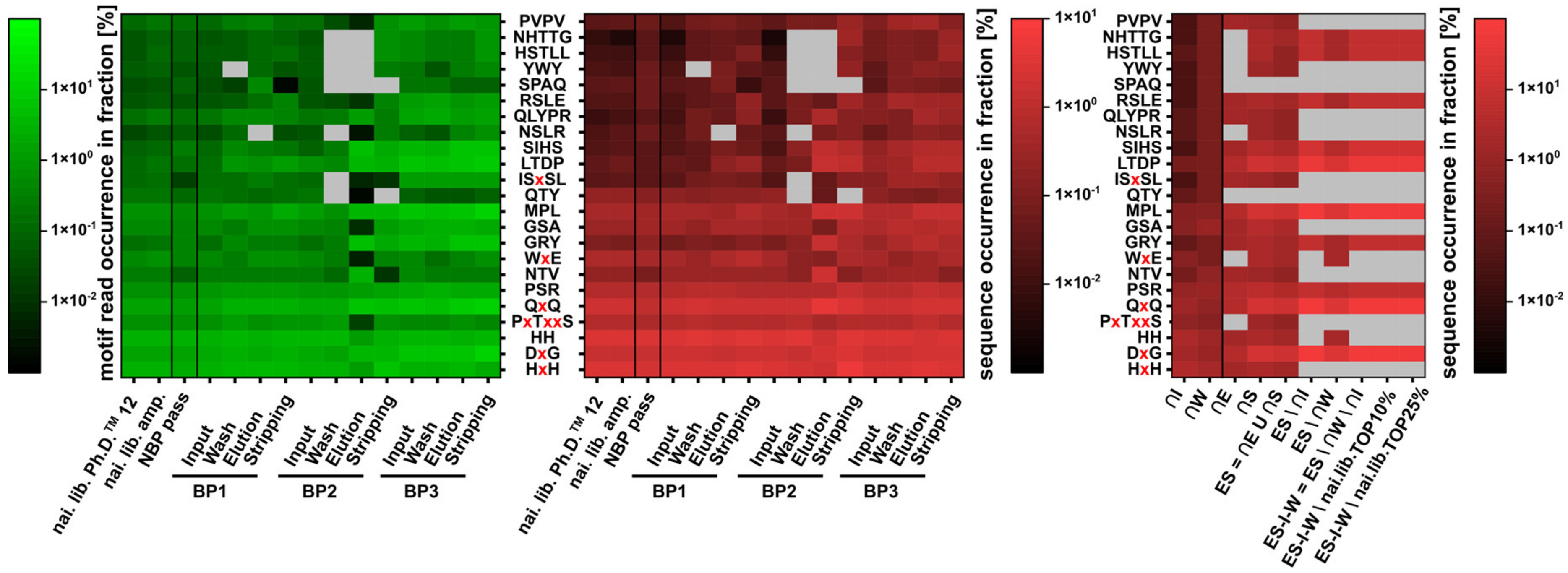
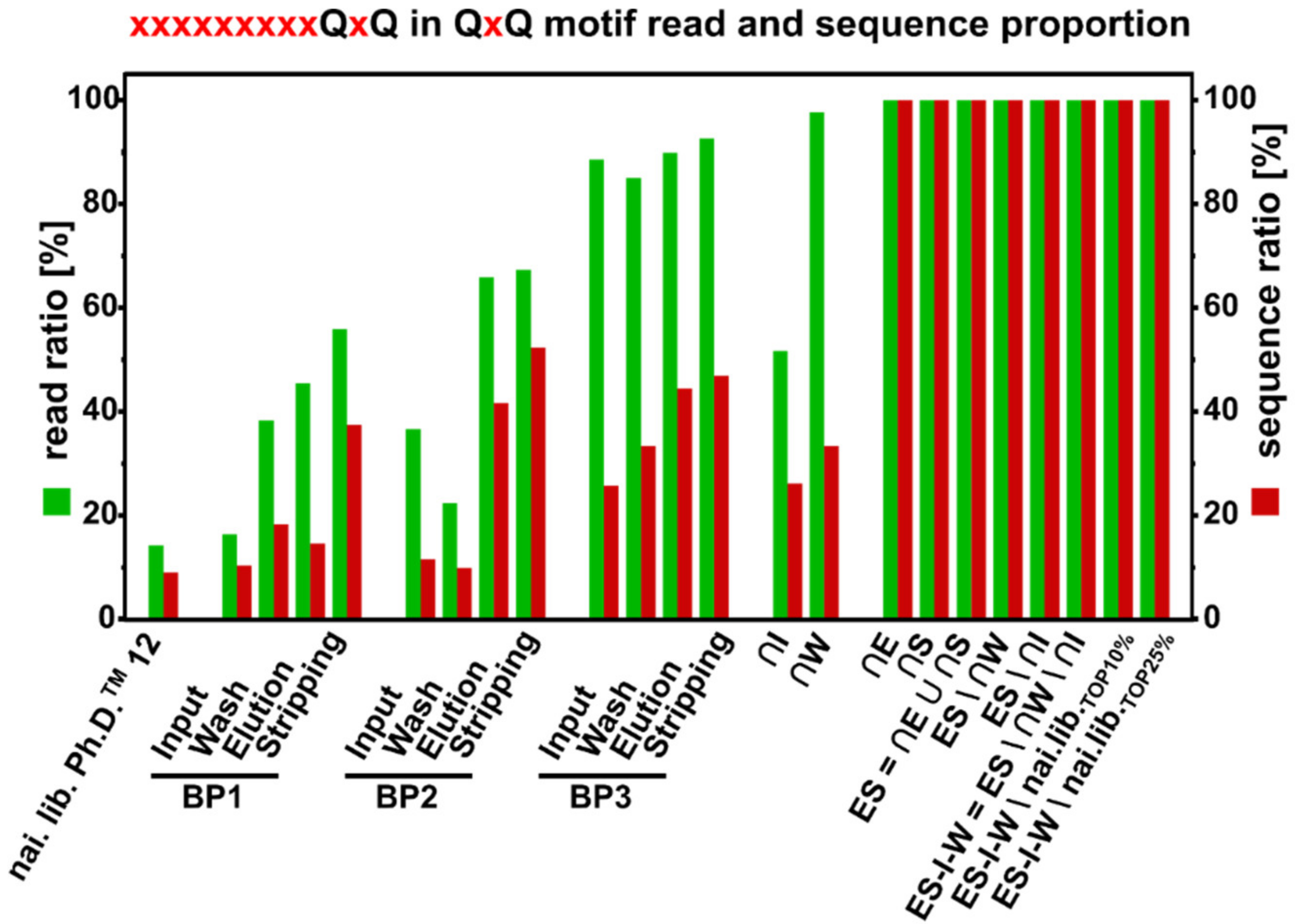
3.6. Core Fraction Calculation
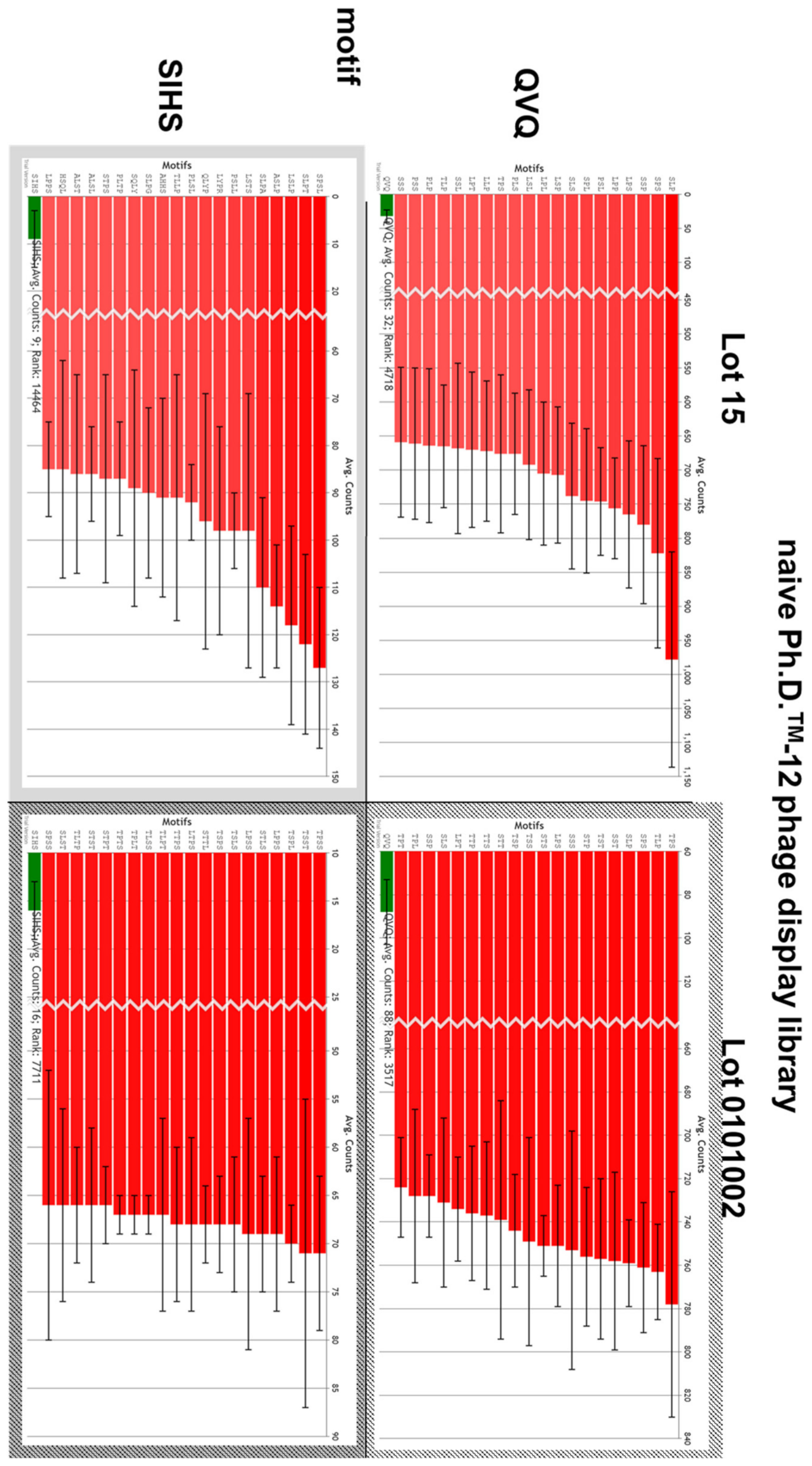
3.7. Sequence Motif Calculation and Comparison
3.8. QxQ Motif
3.9. Motif Comparison with 48 h Discovery Database
4. Discussion
4.1. Comparison of Sanger Sequencing with Next Generation Sequencing
4.2. NGS: Amino Acid Composition
4.3. NGS: Core Fraction Calculation
4.4. NGS: Motif Enrichment
4.5. QxQ Motif: Metal- and Oxyanion Interaction
4.6. QxQ Motif: Biological Occurrence
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Frequency in Naïve Library (%) | |
|---|---|---|
| arginine | R | 5.09 |
| lysine | K | 2.39 |
| aspartic acid | D | 4.00 |
| glutamic acid | E | 2.60 |
| glutamine | Q | 4.14 |
| asparagine | N | 4.65 |
| histidine | H | 5.40 |
| serine | S | 10.80 |
| threonine | T | 9.49 |
| tyrosine | Y | 3.80 |
| cysteine | C | 1.00 |
| tryptophan | W | 1.97 |
| methionine | M | 3.07 |
| alanine | A | 6.88 |
| isoleucine | I | 3.44 |
| leucine | L | 9.13 |
| phenylalanine | F | 2.92 |
| valine | V | 4.97 |
| proline | P | 10.06 |
| glycine | G | 4.21 |



References
- Cullen, W.R. Is Arsenic An Aphrodisiac? Royal Society of Chemistry: Cambridge, UK, 2008. [Google Scholar] [CrossRef]
- Ahuja, S. (Ed.) Arsenic Contamination of Groundwater; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Shen, S.; Li, X.F.; Cullen, W.R.; Weinfeld, M.; Le, X.C. Arsenic binding to proteins. Chem. Rev. 2013, 113, 7769–7792. [Google Scholar] [CrossRef] [PubMed]
- States, J.C. (Ed.) Arsenic: Exposure Sources, Health Risks and Mechanisms of Toxicity; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Yamauchi, H.; Takata, A.; Cao, Y.; Nakamura, K. The Development and Purposes of Arsenic Detoxification Technology; Springer: Singapore, 2019; pp. 199–211. [Google Scholar] [CrossRef]
- Kuzmicheva, G.A.; Belyavskaya, V.A. Peptide phage display in biotechnology and biomedicine. Biochem. Suppl. Ser. B Biomed. Chem. 2017, 11, 1–15. [Google Scholar] [CrossRef]
- Hoen, P.A.T.; Jirka, S.M.; Broeke, B.R.T.; Schultes, E.A.; Aguilera, B.; Pang, K.H.; Heemskerk, H.; Aartsma-Rus, A.; Van Ommen, G.J.; Dunnen, J.T.D. Phage display screening without repetitious selection rounds. Anal. Biochem. 2012, 421, 622–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodnik, M.; Zager, U.; Strukelj, B.; Lunder, M. Phage Display: Selecting Straws Instead of a Needle from a Haystack. Molecules 2011, 16, 790–817. [Google Scholar] [CrossRef] [Green Version]
- Derda, R.; Tang, S.K.Y.; Li, S.C.; Ng, S.; Matochko, W.L.; Jafari, M.R. Diversity of Phage-Displayed Libraries of Peptides during Panning and Amplification. Molecules 2011, 16, 1776–1803. [Google Scholar] [CrossRef] [Green Version]
- Ru, B.; Hoen, P.A.C.T.; Nie, F.; Lin, H.; Guo, F.-B.; Huang, J. PhD7Faster: Predicting clones propagating faster from the Ph.D.-7 Phage Display peptide library. J. Bioinform. Comput. Biol. 2014, 12, 1450005. [Google Scholar] [CrossRef]
- Menendez, A.; Scott, J.K. The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal. Biochem. 2005, 336, 145–157. [Google Scholar] [CrossRef]
- Bakhshinejad, B.; Zade, H.M.; Shekarabi, H.S.Z.; Neman, S. Phage display biopanning and isolation of target-unrelated peptides: In search of nonspecific binders hidden in a combinatorial library. Amino Acids 2016, 48, 2699–2716. [Google Scholar] [CrossRef]
- McIlvaine, T.C. A buffer solution for colorimetric omparison. J. Biol. Chem. 1921, 49, 183–186. [Google Scholar]
- Schönberger, N.; Braun, R.; Matys, S.; Lederer, F.; Lehmann, F.; Flemming, K.; Pollmann, K. Chromatopanning for the identification of gallium binding peptides. J. Chromatogr. A 2019, 1600, 158–166. [Google Scholar] [CrossRef]
- Nian, R.; Kim, D.S.; Nguyen, T.; Tan, L.; Kim, C.-W.; Yoo, I.-K.; Choe, W.-S. Chromatographic biopanning for the selection of peptides with high specificity to Pb2+ from phage displayed peptide library. J. Chromatogr. A 2010, 1217, 5940–5949. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014, 1079, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Shave, S.; Mann, S.; Koszela, J.; Kerr, A.; Auer, M. PuLSE: Quality control and quantification of peptide sequences explored by phage display libraries. PLoS ONE 2018, 13, e0193332. [Google Scholar] [CrossRef] [PubMed]
- Shea, J.P.; Chou, M.F.; Quader, S.A.; Ryan, J.K.; Church, G.M.; Schwartz, D. pLogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods 2013, 10, 1211–1212. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Livingstone, C.D.; Barton, G.J. Protein sequence alignments: A strategy for the hierarchical analysis of residue conservation. Bioinformatics 1993, 9, 745–756. [Google Scholar] [CrossRef] [Green Version]
- Rodi, D.J.; Soares, A.S.; Makowski, L. Quantitative Assessment of Peptide Sequence Diversity in M13 Combinatorial Peptide Phage Display Libraries. J. Mol. Biol. 2002, 322, 1039–1052. [Google Scholar] [CrossRef]
- Derda, R.; Waters, P.; Li, C.; O’Gara, Z. 48Hour Discovery. 2020. Available online: www.48hd.cloud (accessed on 31 August 2020).
- Matochko, W.L.; Li, C.S.; Tang, S.K.Y.; Derda, R. Prospective identification of parasitic sequences in phage display screens. Nucleic Acids Res. 2014, 42, 1784–1798. [Google Scholar] [CrossRef] [Green Version]
- National Research Council. Chemistry and Analysis of Arsenic Species in Water, Food, Urine, Blood, Hair, and Nails. In Arsenic in Drinking Water; National Academies Press: Washington, DC, USA, 1999. [Google Scholar]
- Rodi, D.J.; Makowski, L.; Kay, B.K. One from column A and two from column B: The benefits of phage display in molecular-recognition studies. Curr. Opin. Chem. Biol. 2002, 6, 92–96. [Google Scholar] [CrossRef]
- Kuzmicheva, G.A.; Jayanna, P.K.; Sorokulova, I.B.; Petrenko, V.A. Diversity and censoring of landscape phage libraries. Protein Eng. Des. Sel. 2008, 22, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Matochko, W.L.; Chu, K.; Jin, B.; Lee, S.W.; Whitesides, G.M.; Derda, R. Deep sequencing analysis of phage libraries using Illumina platform. Methods 2012, 58, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Rodi, D.J.; Janes, R.W.; Sanganee, H.J.; Holton, A.R.; Wallace, B.; Makowski, L. Screening of a library of phage-displayed peptides identifies human Bcl-2 as a taxol-binding protein 1 1Edited by I. A. Wilson. J. Mol. Biol. 1999, 285, 197–203. [Google Scholar] [CrossRef]
- Lowman, H.B.; Wells, J.A. Affinity Maturation of Human Growth Hormone by Monovalent Phage Display. J. Mol. Biol. 1993, 234, 564–578. [Google Scholar] [CrossRef]
- Kay, B.K.; Adey, N.B.; Yun-Sheng, H.; Manfredi, J.P.; Mataragnon, A.H.; Fowlkes, D.M. An M13 phage library displaying random 38-amino-acid peptides as a source of novel sequences with affinity to selected targets. Gene 1993, 128, 59–65. [Google Scholar] [CrossRef]
- Nagler, C.; Nagler, G.; Kuhn, A. Cysteine Residues in the Transmembrane Regions of M13 Procoat Protein Suggest that Oligomeric Coat Proteins Assemble onto Phage Progeny. J. Bacteriol. 2007, 189, 2897–2905. [Google Scholar] [CrossRef] [Green Version]
- Yamane, K.; Mizushima, S. Introduction of basic amino acid residues after the signal peptide inhibits protein translocation across the cytoplasmic membrane of Escherichia coli. Relation to the orientation of membrane proteins. J. Biol. Chem. 1988, 263, 19690–19696. [Google Scholar]
- Nilsson, I.; Von Heijne, G. A signal peptide with a proline next to the cleavage site inhibits leader peptidase when present in a sec -independent protein. FEBS Lett. 1992, 299, 243–246. [Google Scholar] [CrossRef] [Green Version]
- Malik, P.; Terry, T.D.; Gowda, L.R.; Langara, A.; Petukhov, S.A.; Symmons, M.F.; Welsh, L.C.; Marvin, D.A.; Perham, R.N. Role of Capsid Structure and Membrane Protein Processing in Determining the Size and Copy Number of Peptides Displayed on the Major Coat Protein of Filamentous Bacteriophage. J. Mol. Biol. 1996, 260, 9–21. [Google Scholar] [CrossRef]
- Zalucki, Y.M.; Jennings, M.P. Signal peptidase I processed secretory signal sequences: Selection for and against specific amino acids at the second position of mature protein. Biochem. Biophys. Res. Commun. 2017, 483, 972–977. [Google Scholar] [CrossRef] [Green Version]
- Choo, K.H.; Ranganathan, S. Flanking signal and mature peptide residues influence signal peptide cleavage. BMC Bioinform. 2008, 9, S15. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimizadeh, W.; Rajabibazl, M. Bacteriophage Vehicles for Phage Display: Biology, Mechanism, and Application. Curr. Microbiol. 2014, 69, 109–120. [Google Scholar] [CrossRef]
- Wilson, D.R.; Finlay, B.B. Phage display: Applications, innovations, and issues in phage and host biology. Can. J. Microbiol. 1998, 44, 313–329. [Google Scholar] [CrossRef]
- Herman, R.E.; Badders, D.; Fuller, M.; Makienko, E.G.; Houston, M.E., Jr.; Quay, S.C.; Johnson, P.H.; Houston, M.E. The Trp Cage Motif as a Scaffold for the Display of a Randomized Peptide Library on Bacteriophage T7. J. Biol. Chem. 2007, 282, 9813–9824. [Google Scholar] [CrossRef] [Green Version]
- Schönberger, N.; Zeitler, C.; Braun, R.; Lederer, F.; Matys, S.; Pollmann, K. Directed Evolution and Engineering of Gallium-Binding Phage Clones-A Preliminary Study. Biomimetics 2019, 4, 35. [Google Scholar] [CrossRef] [Green Version]
- Schönberger, N.; Taylor, C.; Schrader, M.; Drobot, B.; Matys, S.; Lederer, F.L. Gallium-binding peptides as a tool for the sustainable treatment of industrial waste streams. 2020; (under review). [Google Scholar]
- Diatchenko, L.; Lau, Y.F.; Campbell, A.P.; Chenchik, A.; Moqadam, F.; Huang, B.; Lukyanov, S.; Gurskaya, N.; Sverdlov, E.D.; Siebert, P.D. Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc. Natl. Acad. Sci. USA 1996, 93, 6025–6030. [Google Scholar] [CrossRef] [Green Version]
- Rebrikov, D.V.; Desai, S.M.; Siebert, P.D.; Lukyanov, S.A. Suppression Subtractive Hybridization. Gene Expression Profiling; Humana Press: Totowa, NJ, USA, 2004; Volume 258, pp. 107–134. [Google Scholar] [CrossRef]
- Vargas-Sanchez, K.; Vekris, A.; Petry, K.G. DNA Subtraction of In Vivo Selected Phage Repertoires for Efficient Peptide Pathology Biomarker Identification in Neuroinflammation Multiple Sclerosis Model. Biomark. Insights 2016, 11, BMI.S32188. [Google Scholar] [CrossRef]
- Yousef, E.N.; Angel, L.A. Comparison of the pH-dependent formation of His and Cys heptapeptide complexes of nickel(II), copper(II), and zinc(II) as determined by ion mobility-mass spectrometry. J. Mass Spectrom. 2020, 55, e4489. [Google Scholar] [CrossRef]
- Kluska, K.; Adamczyk, J.; Krezel, A. Metal binding properties of zinc fingers with a naturally altered metal binding site. Metallomics 2018, 10, 248–263. [Google Scholar] [CrossRef]
- Ren, D.; Penner, N.A.; Slentz, B.E.; Mirzaei, H.; Regnier, F. Evaluating Immobilized Metal Affinity Chromatography for the Selection of Histidine-Containing Peptides in Comparative Proteomics. J. Proteome Res. 2003, 2, 321–329. [Google Scholar] [CrossRef]
- Yamashita, M.M.; Wesson, L.; Eisenman, G.; Eisenberg, D. Where metal ions bind in proteins. Proc. Natl. Acad. Sci. USA 1990, 87, 5648–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Hu, X.; Zhang, X.; Gao, S.; Ding, C.; Feng, Y.; Bao, W. Identification of metal ion binding sites based on amino acid sequences. PLoS ONE 2017, 12, e0183756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenzweig, A.C. Metallochaperones. Chem. Biol. 2002, 9, 673–677. [Google Scholar] [CrossRef] [Green Version]
- Auld, D.S. Zinc coordination sphere in biochemical zinc sites. BioMetals 2001, 14, 271–313. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Sóvágó, I. Metal complexes of amino acids and peptides, In Amino Acids, Peptides and Proteins; RSC Publishing: Cambridge, UK, 2012; Volume 37, pp. 66–118. [Google Scholar] [CrossRef]
- Kožíšek, M.; Svatoš, A.; Buděšínský, M.; Mück, A.; Bauer, M.C.; Kotrba, P.; Ruml, T.; Havlas, Z.; Linse, S.; Rulíšek, L. Molecular Design of Specific Metal-Binding Peptide Sequences from Protein Fragments: Theory and Experiment. Chem. A Eur. J. 2008, 14, 7836–7846. [Google Scholar] [CrossRef]
- Yang, Y.; Mitri, K.; Zhang, C.; Boysen, R.I.; Hearn, M.C.T.W. Promiscuity of host cell proteins in the purification of histidine tagged recombinant xylanase A by IMAC procedures: A case study with a Ni2+-tacn-based IMAC system. Protein Expr. Purif. 2019, 162, 51–61. [Google Scholar] [CrossRef]
- Yantsevich, A.V.; Dzichenka, Y.V.; Ivanchik, A.V.; Shapiro, M.A.; Trawkina, M.; Shkel, T.V.; Gilep, A.A.; Sergeev, G.V.; Usanov, S.A. Proteomic analysis of contaminants in recombinant membrane hemeproteins expressed in E. coli and isolated by metal affinity chromatography. Appl. Biochem. Microbiol. 2017, 53, 173–186. [Google Scholar] [CrossRef]
- Bush, M.F.; Oomens, J.; Saykally, R.J.; Williams, E.R. Alkali Metal Ion Binding to Glutamine and Glutamine Derivatives Investigated by Infrared Action Spectroscopy and Theory. J. Phys. Chem. A 2008, 112, 8578–8584. [Google Scholar] [CrossRef]
- Neumann, P.Z.; Sass-Kortsak, A. The State of Copper in Human Serum: Evidence for an Amino Acid-bound Fraction. J. Clin. Investig. 1967, 46, 646–658. [Google Scholar] [CrossRef]
- Chiera, N.M.; Rowinska-Zyrek, M.; Wieczorek, R.; Guerrini, R.; Witkowska, D.; Remelli, M.; Henryk, K. Unexpected impact of the number of glutamine residues on metal complex stability. Metallomics 2013, 5, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Cetinel, S.; Dinçer, S.; Cebeci, A.; Oren, E.E.; Whitaker, J.D.; Schwartz, D.T.; Karaguler, N.G.; Sarikaya, M.; Tamerler, C. Peptides to bridge biological-platinum materials interface. Bioinspired Biomim. Nanobiomater. 2012, 1, 143–153. [Google Scholar] [CrossRef]
- Dokmanić, I.; Šikić, M.; Tomić, S. Metals in proteins: Correlation between the metal-ion type, coordination number and the amino-acid residues involved in the coordination. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Barber-Zucker, S.; Shaanan, B.; Zarivach, R. Transition metal binding selectivity in proteins and its correlation with the phylogenomic classification of the cation diffusion facilitator protein family. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsui, K.; Matsumoto, A.; Ohtsuka, S.; Ohtsubo, M.; Yoshimura, A. Cloning and characterization of a novel p21(Cip1/Waf1)-interacting zinc finger protein, Ciz1. Biochem. Biophys. Res. Commun. 1999, 264, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-N.; Yang, L.; Ling, J.-Y.; Czajkowsky, D.M.; Wang, J.-F.; Zhang, X.-W.; Zhou, Y.-M.; Ge, F.; Yang, M.-K.; Xiong, Q.; et al. Systematic identification of arsenic-binding proteins reveals that hexokinase-2 is inhibited by arsenic. Proc. Natl. Acad. Sci. USA 2015, 112, 15084–15089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Demel, S.; Shi, J.; Gladysheva, T.; Gatti, D.L.; Rosen, B.P.; Edwards, B.F. Insights into the Structure, Solvation, and Mechanism of ArsC Arsenate Reductase, a Novel Arsenic Detoxification Enzyme. Structure 2001, 9, 1071–1081. [Google Scholar] [CrossRef] [Green Version]
- Kitchin, K.T.; Wallace, K. Arsenite binding to synthetic peptides based on the Zn finger region and the estrogen binding region of the human estrogen receptor-α. Toxicol. Appl. Pharmacol. 2005, 206, 66–72. [Google Scholar] [CrossRef]
- Rhys, N.H.; Soper, A.K.; Dougan, L. The Hydrogen-Bonding Ability of the Amino Acid Glutamine Revealed by Neutron Diffraction Experiments. J. Phys. Chem. B 2012, 116, 13308–13319. [Google Scholar] [CrossRef]
- Shook, R.L.; Borovik, A.S. Role of the secondary coordination sphere in metal-mediated dioxygen activation. Inorg. Chem. 2010, 49, 3646–3660. [Google Scholar] [CrossRef] [Green Version]
- Krenkel, P.A. (Ed.) Heavy Metals in the Aquatic Environment; Elsevier: Amsterdam, The Netherlands, 1975. [Google Scholar] [CrossRef]
- Boudreaux, D.A.; Chaney, J.; Maiti, T.K.; Das, C. Contribution of active site glutamine to rate enhancement in ubiquitin C-terminal hydrolases. FEBS J. 2012, 279, 1106–1118. [Google Scholar] [CrossRef]
- Warelow, T.P.; Pushie, M.J.; Cotelesage, J.J.H.; Santini, J.M.; George, G.N. The active site structure and catalytic mechanism of arsenite oxidase. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Mukhopadhyay, R.; Rosen, B.P. Identification of a triad of arginine residues in the active site of the ArsC arsenate reductase of plasmid R773. FEMS Microbiol. Lett. 2003, 227, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Ménard, R.; Carrière, J.; Laflamme, P.; Plouffe, C.; Khouri, H.E.; Vernet, T.; Tessier, D.C.; Thomas, D.Y.; Storer, A.C. Contribution of the Glutamine 19 Side Chain to Transition-State Stabilization in the Oxyanion Hole of Papain. Biochemistry 1991, 30, 8924–8928. [Google Scholar] [CrossRef] [PubMed]
- Weidner, E.; Ciesielczyk, F. Removal of Hazardous Oxyanions from the Environment Using Metal-Oxide-Based Materials. Materials 2019, 12, 927. [Google Scholar] [CrossRef] [Green Version]
- Carter, S.L.W.D.E. Arsenate toxicity in human erythrocytes: Characterization of morphologic changes and determination of the mechanism of damage. J. Toxicol. Environ. Health Part A 1998, 53, 345–355. [Google Scholar] [CrossRef]
- Yang, H.-C.; Fu, H.-L.; Lin, Y.-F.; Rosen, B.P. Pathways of Arsenic Uptake and Efflux. In Current Topics in Membranes; Academic Press Inc.: Cambridge, MA, USA, 2012; Volume 69, pp. 325–358. [Google Scholar] [CrossRef] [Green Version]
- Sigrist, C.J.A.; Cerutti, L.; Hulo, N.; Gattiker, A.; Falquet, L.; Pagni, M.; Bairoch, A.; Bucher, P. PROSITE: A documented database using patterns and profiles as motif descriptors. Briefings Bioinform. 2002, 3, 265–274. [Google Scholar] [CrossRef]
- Sigrist, C.J.A.; De Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2012, 41, D344–D347. [Google Scholar] [CrossRef] [Green Version]
- De Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Lee, S.; Lin, X.; Nam, N.H.; Parang, K.; Sun, G. Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc. Natl. Acad. Sci. USA 2003, 100, 14707–14712. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Potts, M.; Kennelly, P.J. The serine, threonine, and/or tyrosine-specific protein kinases and protein phosphatases of prokaryotic organisms: A family portrait. FEMS Microbiol. Rev. 1998, 22, 229–253. [Google Scholar] [CrossRef]
- Silver, S.; Phung, L.T. Genes and enzymes involved in bacterial oxidation and reduction of inorganic arsenic. Appl. Environ. Microbiol. 2005, 71, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Rosen, B.P. Ligand interactions of the ArsC arsenate reductase. J. Biol. Chem. 1997, 272, 21084–21089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinton, L.T.; Bauknight, D.K.; Dasa, S.S.K.; Kelly, K.A. PHASTpep: Analysis Software for Discovery of Cell-Selective Peptides via Phage Display and Next-Generation Sequencing. PLoS ONE 2016, 11, e0155244. [Google Scholar] [CrossRef] [PubMed]
- Vekris, A.; Pilalis, E.; Chatziioannou, A.; Petry, K.G. A Computational Pipeline for the Extraction of Actionable Biological Information From NGS-Phage Display Experiments. Front. Physiol. 2019, 10, 1160. [Google Scholar] [CrossRef]
- Dias-Neto, E.; Nunes, D.N.; Giordano, R.J.; Sun, J.; Botz, G.H.; Yang, K.; Setubal, J.C.; Pasqualini, R.; Pasqualini, R. Next-Generation Phage Display: Integrating and Comparing Available Molecular Tools to Enable Cost-Effective High-Throughput Analysis. PLoS ONE 2009, 4, e8338. [Google Scholar] [CrossRef]
- Huang, J.; Ru, B.; Dai, P. Bioinformatics Resources and Tools for Phage Display. Molecules 2011, 16, 694–709. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Chen, H.; Huang, J. PhD7Faster 2.0: Predicting clones propagating faster from the Ph.D.-7 phage display library by coupling PseAAC and tripeptide composition. PeerJ 2019, 7, e7131. [Google Scholar] [CrossRef]
- He, B.; Chen, H.; Li, N.; Huang, J. Sarotup: A suite of tools for finding potential target-unrelated peptides from phage display data. Int. J. Biol. Sci. 2019, 15, 1452–1459. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Chai, G.; Duan, Y.; Yan, Z.; Qiu, L.; Zhang, H.; Liu, Z.; He, Q.; Han, K.; Ru, B.; et al. BDB: Biopanning data bank. Nucleic Acids Res. 2016, 44, D1127–D1132. [Google Scholar] [CrossRef] [Green Version]



| Peptide Sequence | pI | Occurrence | Peptide Sequence | pI | Occurrence |
|---|---|---|---|---|---|
| FHMPLTDPGQVQ | 5.08 | 11/68 | |||
| SIHSVTKGRYPV | 9.99 | 11/68 | |||
| MKAHHSQLYPRH | 9.99 | 2/68 | |||
| ANGSEYNLLQQS | 4.00 | 1/68 | DFPRTKSETRAP | 8.75 | 1/68 |
| DGMTKPAQHTNR | 8.75 | 1/68 | DPMQKSHLVSQS | 6.74 | 1/68 |
| DVLQPEGLTIPL | 3.67 | 1/68 | EDSGLASEKIAR | 4.68 | 1/68 |
| ERNVTSDDPGSI | 4.03 | 1/68 | FSDRVGSILNSP | 5.84 | 1/68 |
| GAISDYTPSQFY | 3.80 | 1/68 | GSAARTISPSLL | 9.75 | 1/68 |
| GVAAAVSVSNAS | 5.52 | 1/68 | GYLGSYRAHEDS | 5.32 | 1/68 |
| HSPALDRLHGIP | 6.92 | 1/68 | LPITEKEPYDKF | 4.68 | 1/68 |
| LQTYDNPAKSIN | 5.83 | 1/68 | NEVNNSSGAPKQ | 6.00 | 1/68 |
| NLTYKQINPAAF | 8.59 | 1/68 | NNHNGPDVTYWV | 5.08 | 1/68 |
| NYLPHQSSSPSR | 8.75 | 1/68 | QARTAMSLEQHL | 6.75 | 1/68 |
| QCLASCLGPQRV | 8.07 | 1/68 | RISYKPDSWQAS | 8.59 | 1/68 |
| RLPSYTTGLIAN | 8.75 | 1/68 | SMSSGLTSNKSY | 8.31 | 1/68 |
| SDNLHYTLLPMH | 5.92 | 1/68 | SNKNLDTRILTK | 9.99 | 1/68 |
| SHMLSSEWESAS | 4.51 | 1/68 | STNLYNTVAYQD | 3.80 | 1/68 |
| SITELLNAAHST | 5.22 | 1/68 | SYMWATGSPLAY | 5.24 | 1/68 |
| SLSPAGYTRLSL | 8.46 | 1/68 | TGKLIESSPDSI | 4.37 | 1/68 |
| THSEPYYPHSHK | 6.74 | 1/68 | TIKEPFPNRDLY | 5.73 | 1/68 |
| TISAFTSFMPTN | 5.19 | 1/68 | VRPTTEYMETSM | 4.53 | 1/68 |
| WGVTKPIRTSTL | 11.00 | 1/68 | QINQDSLHTPAA | 5.08 | 1/68 |
| YDAIQRPTGQLS | 5.84 | 1/68 | YQRPANLSMEDR | 6.07 | 1/68 |
| Biopanning | Fraction | Reads | Unique Sequences |
| naïve library Ph.D.TM–12 LOT 0151606 | 133,163 | 97,563 | |
| amplification of naïve library | 85,533 | 59,375 | |
| biopanning 1 BP1 | input | 87,883 | 67,705 |
| wash | 5271 | 2915 | |
| elution | 3975 | 2563 | |
| stripping | 124,565 | 16,235 | |
| biopanning 2 BP2 | input | 109,784 | 82,399 |
| wash | 3274 | 1185 | |
| elution | 72,950 | 3536 | |
| stripping | 20,167 | 1999 | |
| biopanning 3 BP3 | input | 74,389 | 20,331 |
| wash | 2001 | 999 | |
| elution | 2373 | 1268 | |
| stripping | 2828 | 1487 |
| Core fFaction (Set) | Read Number | Unique Sequences |
|---|---|---|
| 15,027 | 2912 | |
| 4931 | 381 | |
| 209 | 56 | |
| 5304 | 74 | |
| 1753 | 113 | |
| 613 | 48 | |
| 51 | 14 | |
| 51 | 14 | |
| 51 | 14 | |
| 41 | 13 |
| Fraction | FHMPLTDPGQVQ | SIHSVTKGRYPV | MKAHHSQLYPRH | |||
|---|---|---|---|---|---|---|
| 0 Sanger seq.+ | 11/68 | (16.18%) | 11/68 | (16.18%) | 02/68 | (2.94%) |
| 1 naïve library | 143/143,424 | (0.10%) | 110/143,424 | (0.08%) | 232/143,424 | (0.16%) |
| 2 ampli. naï. lib. | 115/85,533 | (0.13%) | 99/85,533 | (0.12%) | 208/85,533 | (0.24%) |
| 3 input BP1 | 84/87,883 | (0.10%) | 69/87,883 | (0.08%) | 160/87,883 | (0.18%) |
| 4 elution BP3 | 134/2373 | (5.65%) | 147/2373 | (6.19%) | 28/2373 | (1.18%) |
| 5 stripping BP3 | 252/2828 | (8.91%) | 164/2828 | (5.80%) | 41/2828 | (1.45%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, R.; Schönberger, N.; Vinke, S.; Lederer, F.; Kalinowski, J.; Pollmann, K. Application of Next Generation Sequencing (NGS) in Phage Displayed Peptide Selection to Support the Identification of Arsenic-Binding Motifs. Viruses 2020, 12, 1360. https://doi.org/10.3390/v12121360
Braun R, Schönberger N, Vinke S, Lederer F, Kalinowski J, Pollmann K. Application of Next Generation Sequencing (NGS) in Phage Displayed Peptide Selection to Support the Identification of Arsenic-Binding Motifs. Viruses. 2020; 12(12):1360. https://doi.org/10.3390/v12121360
Chicago/Turabian StyleBraun, Robert, Nora Schönberger, Svenja Vinke, Franziska Lederer, Jörn Kalinowski, and Katrin Pollmann. 2020. "Application of Next Generation Sequencing (NGS) in Phage Displayed Peptide Selection to Support the Identification of Arsenic-Binding Motifs" Viruses 12, no. 12: 1360. https://doi.org/10.3390/v12121360