Differences in HIV Markers between Infected Individuals Treated with Different ART Regimens: Implications for the Persistence of Viral Reservoirs


Abstract
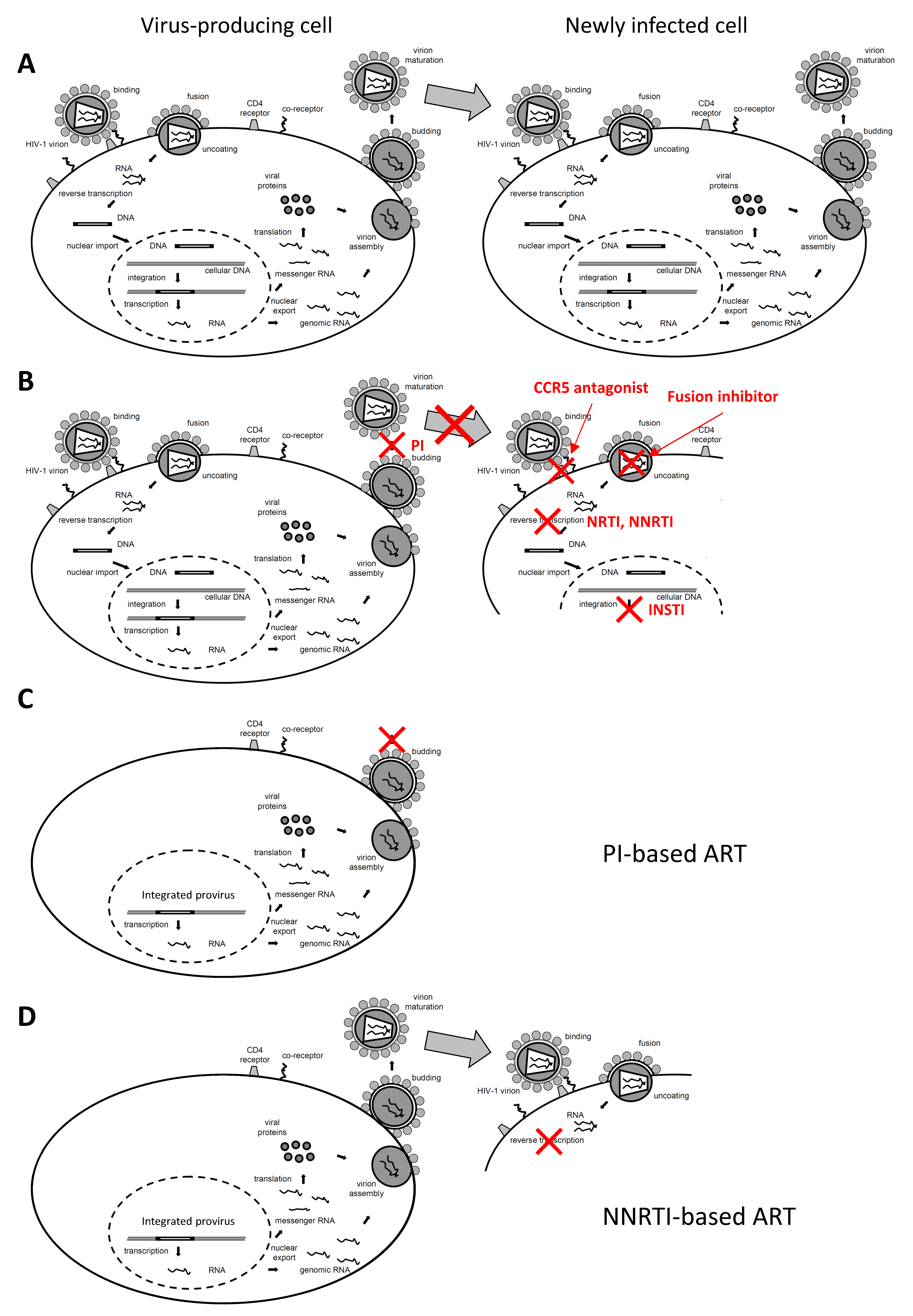
:1. Introduction
2. Persistence of HIV Reservoirs on ART
3. Sources of HIV Residual Viremia on ART
4. Impact of ART Intensification on Residual Viremia and Other HIV Reservoir Markers
5. Differences in Residual Viremia and Other HIV Reservoir Markers between Infected Individuals Treated with Different ART Regimens
6. Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Deeks, S.G.; Lewin, S.R.; Havlir, D.V. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Sebaaly, J.C.; Kelley, D. Single-Tablet Regimens for the Treatment of HIV-1 Infection. Ann. Pharm. 2017, 51, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.; Darcis, G.; Lee, M.Y.; Bouchat, S.; Van Driessche, B.; Purcell, D.F.J.; Van Lint, C. The Molecular Biology of HIV Latency. Adv. Exp. Med. Biol. 2018, 1075, 187–212. [Google Scholar] [CrossRef]
- Pasternak, A.O.; Berkhout, B. HIV Reservoir: Finding the Right Needles in a Needlestack. Cell Host Microbe 2016, 20, 280–282. [Google Scholar] [CrossRef] [Green Version]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef]
- Dimmock, N.; Easton, A.; Leppard, K.N. Introduction to Modern Virology, 6th ed.; Blackwell Publishing: Malden, MA, USA, 2007. [Google Scholar]
- Fischer, M.; Wong, J.K.; Russenberger, D.; Joos, B.; Opravil, M.; Hirschel, B.; Trkola, A.; Kuster, H.; Weber, R.; Gunthard, H.F. Residual cell-associated unspliced HIV-1 RNA in peripheral blood of patients on potent antiretroviral therapy represents intracellular transcripts. Antivir. Ther. 2002, 7, 91–103. [Google Scholar]
- Pasternak, A.O.; de Bruin, M.; Jurriaans, S.; Bakker, M.; Berkhout, B.; Prins, J.M.; Lukashov, V.V. Modest nonadherence to antiretroviral therapy promotes residual HIV-1 replication in the absence of virological rebound in plasma. J. Infect. Dis. 2012, 206, 1443–1452. [Google Scholar] [CrossRef]
- Pasternak, A.O.; Jurriaans, S.; Bakker, M.; Prins, J.M.; Berkhout, B.; Lukashov, V.V. Cellular levels of HIV unspliced RNA from patients on combination antiretroviral therapy with undetectable plasma viremia predict the therapy outcome. PLOS ONE 2009, 4, e8490. [Google Scholar] [CrossRef]
- DeMaster, L.K.; Liu, X.H.; VanBelzen, D.J.; Trinite, B.; Zheng, L.J.; Agosto, L.M.; Migueles, S.A.; Connors, M.; Sambucetti, L.; Levy, D.N.; et al. A Subset of CD4/CD8 Double-Negative T Cells Expresses HIV Proteins in Patients on Antiretroviral Therapy. J. Virol. 2016, 90, 2165–2179. [Google Scholar] [CrossRef] [Green Version]
- Baxter, A.E.; Niessl, J.; Fromentin, R.; Richard, J.; Porichis, F.; Charlebois, R.; Massanella, M.; Brassard, N.; Alsahafi, N.; Delgado, G.G.; et al. Single-Cell Characterization of Viral Translation-Competent Reservoirs in HIV-Infected Individuals. Cell Host Microbe 2016, 20, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Sarracino, A.; Gharu, L.; Kula, A.; Pasternak, A.O.; Avettand-Fenoel, V.; Rouzioux, C.; Bardina, M.; De Wit, S.; Benkirane, M.; Berkhout, B.; et al. Posttranscriptional Regulation of HIV-1 Gene Expression during Replication and Reactivation from Latency by Nuclear Matrix Protein MATR3. MBio 2018, 9, e02158-18. [Google Scholar] [CrossRef] [Green Version]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD4+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef] [Green Version]
- Pardons, M.; Baxter, A.E.; Massanella, M.; Pagliuzza, A.; Fromentin, R.; Dufour, C.; Leyre, L.; Routy, J.P.; Kaufmann, D.E.; Chomont, N. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLOS Pathog. 2019, 15, e1007619. [Google Scholar] [CrossRef]
- Pace, M.J.; Agosto, L.; Graf, E.H.; O’Doherty, U. HIV reservoirs and latency models. Virology 2011, 411, 344–354. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, A.O.; Berkhout, B. What do we measure when we measure cell-associated HIV RNA. Retrovirology 2018, 15, 13. [Google Scholar] [CrossRef]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Hiener, B.; Horsburgh, B.A.; Eden, J.S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4(+) T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Pinzone, M.R.; VanBelzen, D.J.; Weissman, S.; Bertuccio, M.P.; Cannon, L.; Venanzi-Rullo, E.; Migueles, S.; Jones, R.B.; Mota, T.; Joseph, S.B.; et al. Longitudinal HIV sequencing reveals reservoir expression leading to decay which is obscured by clonal expansion. Nat. Commun. 2019, 10, 728. [Google Scholar] [CrossRef]
- Peluso, M.J.; Bacchetti, P.; Ritter, K.D.; Beg, S.; Lai, J.; Martin, J.N.; Hunt, P.W.; Henrich, T.J.; Siliciano, J.D.; Siliciano, R.F.; et al. Differential decay of intact and defective proviral DNA in HIV-1-infected individuals on suppressive antiretroviral therapy. JCI Insight 2020, 5, 132997. [Google Scholar] [CrossRef] [Green Version]
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Finzi, D.; Plaeger, S.F.; Dieffenbach, C.W. Defective virus drives human immunodeficiency virus infection, persistence, and pathogenesis. Clin. Vaccine Immunol. 2006, 13, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-Based Measures of Viral Persistence Are Associated With Immune Activation and Programmed Cell Death Protein 1 (PD-1)-Expressing CD4+ T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef] [Green Version]
- Baxter, A.E.; O’Doherty, U.; Kaufmann, D.E. Beyond the replication-competent HIV reservoir: Transcription and translation-competent reservoirs. Retrovirology 2018, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.S.; Capoferri, A.A.; Beg, S.A.; Huang, S.H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 2017, 21, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Avettand-Fènoël, V.; Hocqueloux, L.; Ghosn, J.; Cheret, A.; Frange, P.; Melard, A.; Viard, J.P.; Rouzioux, C. Total HIV-1 DNA, a Marker of Viral Reservoir Dynamics with Clinical Implications. Clin. Microbiol. Rev. 2016, 29, 859–880. [Google Scholar] [CrossRef] [Green Version]
- Imamichi, H.; Smith, M.; Adelsberger, J.W.; Izumi, T.; Scrimieri, F.; Sherman, B.T.; Rehm, C.A.; Imamichi, T.; Pau, A.; Catalfamo, M.; et al. Defective HIV-1 proviruses produce viral proteins. Proc. Natl. Acad. Sci. USA 2020, 117, 3704–3710. [Google Scholar] [CrossRef] [Green Version]
- Zicari, S.; Sessa, L.; Cotugno, N.; Ruggiero, A.; Morrocchi, E.; Concato, C.; Rocca, S.; Zangari, P.; Manno, E.C.; Palma, P. Immune Activation, Inflammation, and Non-AIDS Co-Morbidities in HIV-Infected Patients under Long-Term ART. Viruses 2019, 11, 200. [Google Scholar] [CrossRef] [Green Version]
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77. [Google Scholar] [CrossRef]
- Colby, D.J.; Trautmann, L.; Pinyakorn, S.; Leyre, L.; Pagliuzza, A.; Kroon, E.; Rolland, M.; Takata, H.; Buranapraditkun, S.; Intasan, J.; et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med. 2018, 24, 923–926. [Google Scholar] [CrossRef]
- Brodin, J.; Zanini, F.; Thebo, L.; Lanz, C.; Bratt, G.; Neher, R.A.; Albert, J. Establishment and stability of the latent HIV-1 DNA reservoir. Elife 2016, 5, e18889. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, M.R.; Joseph, S.B.; Garrett, N.; Tyers, L.; Moeser, M.; Archin, N.; Council, O.D.; Matten, D.; Zhou, S.; Doolabh, D.; et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med. 2019, 11, eaaw5589. [Google Scholar] [CrossRef] [PubMed]
- Goonetilleke, N.; Clutton, G.; Swanstrom, R.; Joseph, S.B. Blocking Formation of the Stable HIV Reservoir: A New Perspective for HIV-1 Cure. Front. Immunol. 2019, 10, 1966. [Google Scholar] [CrossRef] [Green Version]
- Pankau, M.D.; Reeves, D.B.; Harkins, E.; Ronen, K.; Jaoko, W.; Mandaliya, K.; Graham, S.M.; McClelland, R.S.; Matsen Iv, F.A.; Schiffer, J.T.; et al. Dynamics of HIV DNA reservoir seeding in a cohort of superinfected Kenyan women. PLoS Pathog. 2020, 16, e1008286. [Google Scholar] [CrossRef] [PubMed]
- Besson, G.J.; Lalama, C.M.; Bosch, R.J.; Gandhi, R.T.; Bedison, M.A.; Aga, E.; Riddler, S.A.; McMahon, D.K.; Hong, F.; Mellors, J.W. HIV-1 DNA decay dynamics in blood during more than a decade of suppressive antiretroviral therapy. Clin. Infect. Dis. 2014, 59, 1312–1321. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, N.; von Siebenthal, C.; Vongrad, V.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Fellay, J.; Roth, V.; Kok, Y.L.; et al. Determinants of HIV-1 reservoir size and long-term dynamics during suppressive ART. Nat. Commun. 2019, 10, 3193. [Google Scholar] [CrossRef] [Green Version]
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Garcia, J.V. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [Green Version]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef]
- Gras, G.; Kaul, M. Molecular mechanisms of neuroinvasion by monocytes-macrophages in HIV-1 infection. Retrovirology 2010, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Yukl, S.A.; Shergill, A.K.; Ho, T.; Killian, M.; Girling, V.; Epling, L.; Li, P.; Wong, L.K.; Crouch, P.; Deeks, S.G.; et al. The distribution of HIV DNA and RNA in cell subsets differs in gut and blood of HIV-positive patients on ART: Implications for viral persistence. J. Infect. Dis. 2013, 208, 1212–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenabian, M.A.; Costiniuk, C.T.; Mehraj, V.; Ghazawi, F.M.; Fromentin, R.; Brousseau, J.; Brassard, P.; Bélanger, M.; Ancuta, P.; Bendayan, R.; et al. Immune tolerance properties of the testicular tissue as a viral sanctuary site in ART-treated HIV-infected adults. AIDS 2016, 30, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Coombs, R.W.; Van Lint, C. Exploring the anatomical HIV reservoirs: Role of the testicular tissue. AIDS 2016, 30, 2891–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcelin, A.G.; Tubiana, R.; Lambert-Niclot, S.; Lefebvre, G.; Dominguez, S.; Bonmarchand, M.; Vauthier-Brouzes, D.; Marguet, F.; Mousset-Simeon, N.; Peytavin, G.; et al. Detection of HIV-1 RNA in seminal plasma samples from treated patients with undetectable HIV-1 RNA in blood plasma. AIDS 2008, 22, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Berkhout, B.; Pasternak, A.O. The Quest for Cellular Markers of HIV Reservoirs: Any Color You Like. Front. Immunol. 2019, 10, 2251. [Google Scholar] [CrossRef] [PubMed]
- Doyle, T.; Smith, C.; Vitiello, P.; Cambiano, V.; Johnson, M.; Owen, A.; Phillips, A.N.; Geretti, A.M. Plasma HIV-1 RNA detection below 50 copies/mL and risk of virologic rebound in patients receiving highly active antiretroviral therapy. Clin. Infect. Dis. 2012, 54, 724–732. [Google Scholar] [CrossRef]
- Dornadula, G.; Zhang, H.; VanUitert, B.; Stern, J.; Livornese, L., Jr.; Ingerman, M.J.; Witek, J.; Kedanis, R.J.; Natkin, J.; DeSimone, J.; et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 1999, 282, 1627–1632. [Google Scholar] [CrossRef] [Green Version]
- Havlir, D.V.; Bassett, R.; Levitan, D.; Gilbert, P.; Tebas, P.; Collier, A.C.; Hirsch, M.S.; Ignacio, C.; Condra, J.; Gunthard, H.F.; et al. Prevalence and predictive value of intermittent viremia with combination hiv therapy. JAMA 2001, 286, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.; Wiegand, A.P.; Maldarelli, F.; Bazmi, H.; Mican, J.M.; Polis, M.; Dewar, R.L.; Planta, A.; Liu, S.; Metcalf, J.A.; et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J. Clin. Microbiol. 2003, 41, 4531–4536. [Google Scholar] [CrossRef] [Green Version]
- Palmisano, L.; Giuliano, M.; Nicastri, E.; Pirillo, M.F.; Andreotti, M.; Galluzzo, C.M.; Bucciardini, R.; Fragola, V.; Andreoni, M.; Vella, S. Residual viraemia in subjects with chronic HIV infection and viral load <50 copies/mL: The impact of highly active antiretroviral therapy. AIDS 2005, 19, 1843–1847. [Google Scholar] [CrossRef]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.J.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cillo, A.R.; Vagratian, D.; Bedison, M.A.; Anderson, E.M.; Kearney, M.F.; Fyne, E.; Koontz, D.; Coffin, J.M.; Piatak, M.; Mellors, J.W. Improved single-copy assays for quantification of persistent HIV-1 viremia in patients on suppressive antiretroviral therapy. J. Clin. Microbiol. 2014, 52, 3944–3951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosiano, M.A.; Jacobs, J.L.; Shutt, K.A.; Cyktor, J.C.; Mellors, J.W. A Simpler and More Sensitive Single-Copy HIV-1 RNA Assay for Quantification of Persistent HIV-1 Viremia in Individuals on Suppressive Antiretroviral Therapy. J. Clin. Microbiol. 2019, 57, e01714–e01718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Siliciano, R.F. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J. Allergy Clin. Immunol. 2008, 122, 22–28. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Siliciano, R.F. Biomarkers of HIV replication. Curr. Opin. HIV AIDS 2010, 5, 491–497. [Google Scholar] [CrossRef]
- Doyle, T.; Geretti, A.M. Low-level viraemia on HAART: Significance and management. Curr. Opin. Infect. Dis. 2012, 25, 17–25. [Google Scholar] [CrossRef]
- Hilldorfer, B.B.; Cillo, A.R.; Besson, G.J.; Bedison, M.A.; Mellors, J.W. New tools for quantifying HIV-1 reservoirs: Plasma RNA single copy assays and beyond. Curr. HIV AIDS Rep. 2012, 9, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S. Advances in detection and monitoring of plasma viremia in HIV-infected individuals receiving antiretroviral therapy. Curr. Opin. HIV AIDS 2013, 8, 87–92. [Google Scholar] [CrossRef]
- Sahu, G.K. Potential implication of residual viremia in patients on effective antiretroviral therapy. AIDS Res. Hum. Retrovir. 2015, 31, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Sarmati, L.; D’Ettorre, G.; Parisi, S.G.; Andreoni, M. HIV Replication at Low Copy Number and its Correlation with the HIV Reservoir: A Clinical Perspective. Curr. HIV Res. 2015, 13, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Q.; Palmer, S. Single-molecule techniques to quantify and genetically characterise persistent HIV. Retrovirology 2018, 15, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.L.; Halvas, E.K.; Tosiano, M.A.; Mellors, J.W. Persistent HIV-1 Viremia on Antiretroviral Therapy: Measurement and Mechanisms. Front. Microbiol. 2019, 10, 2383. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.M.; Perelson, A.S. Residual Viremia in Treated HIV+ Individuals. PLoS Comput. Biol. 2016, 12, e1004677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Scheerder, M.A.; Vrancken, B.; Dellicour, S.; Schlub, T.; Lee, E.; Shao, W.; Rutsaert, S.; Verhofstede, C.; Kerre, T.; Malfait, T.; et al. HIV Rebound Is Predominantly Fueled by Genetically Identical Viral Expansions from Diverse Reservoirs. Cell Host Microbe 2019, 26, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef] [Green Version]
- Maldarelli, F.; Palmer, S.; King, M.S.; Wiegand, A.; Polis, M.A.; Mican, J.; Kovacs, J.A.; Davey, R.T.; Rock-Kress, D.; Dewar, R.; et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 2007, 3, e46. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Picado, J.; Deeks, S.G. Persistent HIV-1 replication during antiretroviral therapy. Curr. Opin. HIV AIDS 2016, 11, 417–423. [Google Scholar] [CrossRef]
- Estes, J.D.; Kityo, C.; Ssali, F.; Swainson, L.; Makamdop, K.N.; Del Prete, G.Q.; Deeks, S.G.; Luciw, P.A.; Chipman, J.G.; Beilman, G.J.; et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 2017, 23, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Rothenberger, M.; Nganou-Makamdop, K.; Kityo, C.; Ssali, F.; Chipman, J.G.; Beilman, G.J.; Hoskuldsson, T.; Anderson, J.; Jasurda, J.; Schmidt, T.E.; et al. Impact of Integrase Inhibition compared to non-nucleoside inhibition on HIV reservoirs in Lymphoid Tissues. J. Acquir. Immune Defic. Syndr. 2019, 81, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Redondo, R.; Fryer, H.R.; Bedford, T.; Kim, E.Y.; Archer, J.; Pond, S.L.K.; Chung, Y.S.; Penugonda, S.; Chipman, J.; Fletcher, C.V.; et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, C.V.; Staskus, K.; Wietgrefe, S.W.; Rothenberger, M.; Reilly, C.; Chipman, J.G.; Beilman, G.J.; Khoruts, A.; Thorkelson, A.; Schmidt, T.E.; et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darcis, G.; Moutschen, M. The effect of treatment simplification on HIV reservoirs. Lancet HIV 2017, 4, e328–e329. [Google Scholar] [CrossRef]
- Di Mascio, M.; Srinivasula, S.; Bhattacharjee, A.; Cheng, L.; Martiniova, L.; Herscovitch, P.; Lertora, J.; Kiesewetter, D. Antiretroviral tissue kinetics: In vivo imaging using positron emission tomography. Antimicrob. Agents Chemother. 2009, 53, 4086–4095. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Telwatte, S.; Hatano, H.; Kashuba, A.D.M.; Cottrell, M.L.; Hoh, R.; Liegler, T.J.; Stephenson, S.; Somsouk, M.; Hunt, P.W.; et al. Antiretroviral Therapy Concentrations Differ in Gut vs. Lymph Node Tissues and Are Associated With HIV Viral Transcription by a Novel RT-ddPCR Assay. J. Acquir. Immune Defic. Syndr. 2020, 83, 530–537. [Google Scholar] [CrossRef]
- Kieffer, T.L.; Finucane, M.M.; Nettles, R.E.; Quinn, T.C.; Broman, K.W.; Ray, S.C.; Persaud, D.; Siliciano, R.F. Genotypic analysis of HIV-1 drug resistance at the limit of detection: Virus production without evolution in treated adults with undetectable HIV loads. J. Infect. Dis. 2004, 189, 1452–1465. [Google Scholar] [CrossRef]
- Bailey, J.R.; Sedaghat, A.R.; Kieffer, T.; Brennan, T.; Lee, P.K.; Wind-Rotolo, M.; Haggerty, C.M.; Kamireddi, A.R.; Liu, Y.; Lee, J.; et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J. Virol. 2006, 80, 6441–6457. [Google Scholar] [CrossRef] [Green Version]
- Joos, B.; Fischer, M.; Kuster, H.; Pillai, S.K.; Wong, J.K.; Boni, J.; Hirschel, B.; Weber, R.; Trkola, A.; Gunthard, H.F. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16725–16730. [Google Scholar] [CrossRef] [Green Version]
- Evering, T.H.; Mehandru, S.; Racz, P.; Tenner-Racz, K.; Poles, M.A.; Figueroa, A.; Mohri, H.; Markowitz, M. Absence of HIV-1 evolution in the gut-associated lymphoid tissue from patients on combination antiviral therapy initiated during primary infection. PLOS. Pathog. 2012, 8, e1002506. [Google Scholar] [CrossRef] [Green Version]
- Josefsson, L.; von, S.S.; Faria, N.R.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Tan, A.; Ho, T.; Lemey, P.; et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc. Natl. Acad. Sci. USA 2013, 110, E4987–E4996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, M.F.; Spindler, J.; Shao, W.; Yu, S.; Anderson, E.M.; O’Shea, A.; Rehm, C.; Poethke, C.; Kovacs, N.; Mellors, J.W.; et al. Lack of detectable HIV-1 molecular evolution during suppressive antiretroviral therapy. PLOS Pathog. 2014, 10, e1004010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zyl, G.U.; Katusiime, M.G.; Wiegand, A.; McManus, W.R.; Bale, M.J.; Halvas, E.K.; Luke, B.; Boltz, V.F.; Spindler, J.; Laughton, B.; et al. No evidence of HIV replication in children on antiretroviral therapy. J. Clin. Investig. 2017, 127, 3827–3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzi, G.; Simonetti, F.R.; Watters, S.A.; Anderson, E.M.; Gouzoulis, M.; Kearney, M.F.; Rote, P.; Lange, C.; Shao, W.; Gorelick, R.; et al. No evidence of ongoing HIV replication or compartmentalization in tissues during combination antiretroviral therapy: Implications for HIV eradication. Sci. Adv. 2019, 5, eaav2045. [Google Scholar] [CrossRef] [Green Version]
- van Zyl, G.; Bale, M.J.; Kearney, M.F. HIV evolution and diversity in ART-treated patients. Retrovirology 2018, 15, 14. [Google Scholar] [CrossRef]
- Kearney, M.F.; Wiegand, A.; Shao, W.; McManus, W.R.; Bale, M.J.; Luke, B.; Maldarelli, F.; Mellors, J.W.; Coffin, J.M. Ongoing HIV Replication During ART Reconsidered. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2017; Volume 4. [Google Scholar]
- Rosenbloom, D.I.S.; Hill, A.L.; Laskey, S.B.; Siliciano, R.F. Re-evaluating evolution in the HIV reservoir. Nature 2017, 551, E6–E9. [Google Scholar] [CrossRef]
- Sigal, A.; Kim, J.T.; Balazs, A.B.; Dekel, E.; Mayo, A.; Milo, R.; Baltimore, D. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 2011, 477, 95–98. [Google Scholar] [CrossRef]
- Pau, A.K.; George, J.M. Antiretroviral therapy: Current drugs. Infect. Dis. Clin. N. Am. 2014, 28, 371–402. [Google Scholar] [CrossRef] [Green Version]
- Dinoso, J.B.; Kim, S.Y.; Wiegand, A.M.; Palmer, S.E.; Gange, S.J.; Cranmer, L.; O’Shea, A.; Callender, M.; Spivak, A.; Brennan, T.; et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 9403–9408. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, R.T.; Zheng, L.; Bosch, R.J.; Chan, E.S.; Margolis, D.M.; Read, S.; Kallungal, B.; Palmer, S.; Medvik, K.; Lederman, M.M.; et al. The effect of raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: A randomized controlled trial. PLOS. Med. 2010, 7, e1000321. [Google Scholar] [CrossRef] [Green Version]
- McMahon, D.; Jones, J.; Wiegand, A.; Gange, S.J.; Kearney, M.; Palmer, S.; McNulty, S.; Metcalf, J.A.; Acosta, E.; Rehm, C.; et al. Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy. Clin. Infect. Dis. 2010, 50, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Buzon, M.J.; Massanella, M.; Llibre, J.M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Gatell, J.M.; Domingo, P.; Paredes, R.; Sharkey, M.; et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat. Med. 2010, 16, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Shergill, A.K.; McQuaid, K.; Gianella, S.; Lampiris, H.; Hare, C.B.; Pandori, M.; Sinclair, E.; Gunthard, H.F.; Fischer, M.; et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 2010, 24, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Hatano, H.; Strain, M.C.; Scherzer, R.; Bacchetti, P.; Wentworth, D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Neaton, J.D.; Tracy, R.P.; et al. Increase in 2-long terminal repeat circles and decrease in D-dimer after raltegravir intensification in patients with treated HIV infection: A randomized, placebo-controlled trial. J. Infect. Dis. 2013, 208, 1436–1442. [Google Scholar] [CrossRef] [Green Version]
- Hatano, H.; Hayes, T.L.; Dahl, V.; Sinclair, E.; Lee, T.H.; Hoh, R.; Lampiris, H.; Hunt, P.W.; Palmer, S.; McCune, J.M.; et al. A randomized, controlled trial of raltegravir intensification in antiretroviral-treated, HIV-infected patients with a suboptimal CD4+ T cell response. J. Infect. Dis. 2011, 203, 960–968. [Google Scholar] [CrossRef]
- Chaillon, A.; Gianella, S.; Lada, S.M.; Perez-Santiago, J.; Jordan, P.; Ignacio, C.; Karris, M.; Richman, D.D.; Mehta, S.R.; Little, S.J.; et al. Size, Composition, and Evolution of HIV DNA Populations during Early Antiretroviral Therapy and Intensification with Maraviroc. J. Virol. 2018, 92, e01589–e01617. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, T.A.; McMahon, J.H.; Chang, J.J.; Audsley, J.; Rhodes, A.; Tennakoon, S.; Dantanarayana, A.; Spelman, T.; Schmidt, T.; Kent, S.J.; et al. The effect of antiretroviral intensification with dolutegravir on residual virus replication in HIV-infected individuals: A randomised, placebo-controlled, double-blind trial. Lancet HIV 2018, 5, e221–e230. [Google Scholar] [CrossRef]
- Henrich, T.J. Dolutegravir intensification and HIV persistence: 3 + 1 = 3. Lancet HIV 2018, 5, e201–e202. [Google Scholar] [CrossRef]
- Puertas, M.C.; Gómez-Mora, E.; Santos, J.R.; Moltó, J.; Urrea, V.; Morón-López, S.; Hernández-Rodríguez, A.; Marfil, S.; Martínez-Bonet, M.; Matas, L.; et al. Impact of intensification with raltegravir on HIV-1-infected individuals receiving monotherapy with boosted PIs. J. Antimicrob. Chemother. 2018, 73, 1940–1948. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, M.; Triques, K.; Kuritzkes, D.R.; Stevenson, M. In vivo evidence for instability of episomal human immunodeficiency virus type 1 cDNA. J. Virol. 2005, 79, 5203–5210. [Google Scholar] [CrossRef] [Green Version]
- Sharkey, M.E.; Teo, I.; Greenough, T.; Sharova, N.; Luzuriaga, K.; Sullivan, J.L.; Bucy, R.P.; Kostrikis, L.G.; Haase, A.; Veryard, C.; et al. Persistence of episomal HIV-1 infection intermediates in patients on highly active anti-retroviral therapy. Nat. Med. 2000, 6, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Kieffer, T.L.; Ruff, C.T.; Buck, C.; Gange, S.J.; Siliciano, R.F. Intrinsic stability of episomal circles formed during human immunodeficiency virus type 1 replication. J. Virol. 2002, 76, 4138–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, S.L.; Johnson, E.P.; Bushman, F.D. Human immunodeficiency virus cDNA metabolism: Notable stability of two-long terminal repeat circles. J. Virol. 2002, 76, 3739–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, M.J.; Graf, E.H.; O’Doherty, U. HIV 2-long terminal repeat circular DNA is stable in primary CD4+T Cells. Virology 2013, 441, 18–21. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Picado, J.; Zurakowski, R.; Buzón, M.J.; Stevenson, M. Episomal HIV-1 DNA and its relationship to other markers of HIV-1 persistence. Retrovirology 2018, 15, 15. [Google Scholar] [CrossRef] [Green Version]
- Besson, G.J.; McMahon, D.; Maldarelli, F.; Mellors, J.W. Short-course raltegravir intensification does not increase 2 long terminal repeat episomal HIV-1 DNA in patients on effective antiretroviral therapy. Clin. Infect. Dis. 2012, 54, 451–453. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Coombs, R.W.; Chan, E.S.; Bosch, R.J.; Zheng, L.; Margolis, D.M.; Read, S.; Kallungal, B.; Chang, M.; Goecker, E.A.; et al. No effect of raltegravir intensification on viral replication markers in the blood of HIV-1-infected patients receiving antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2012, 59, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Vallejo, A.; Gutierrez, C.; Hernandez-Novoa, B.; Diaz, L.; Madrid, N.; Abad-Fernandez, M.; Dronda, F.; Perez-Elias, M.J.; Zamora, J.; Muñoz, E.; et al. The effect of intensification with raltegravir on the HIV-1 reservoir of latently infected memory CD4 T cells in suppressed patients. AIDS 2012, 26, 1885–1894. [Google Scholar] [CrossRef]
- Puertas, M.C.; Noguera-Julian, M.; Massanella, M.; Pou, C.; Buzon, M.J.; Clotet, B.; Stevenson, M.; Paredes, R.; Blanco, J.; Martinez-Picado, J. Lack of concordance between residual viremia and viral variants driving de novo infection of CD4(+) T cells on ART. Retrovirology 2016, 13, 51. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, C.; Diaz, L.; Vallejo, A.; Hernandez-Novoa, B.; Abad, M.; Madrid, N.; Dahl, V.; Rubio, R.; Moreno, A.M.; Dronda, F.; et al. Intensification of antiretroviral therapy with a CCR5 antagonist in patients with chronic HIV-1 infection: Effect on T cells latently infected. PLOS ONE 2011, 6, e27864. [Google Scholar] [CrossRef]
- Madrid-Elena, N.; García-Bermejo, M.L.; Serrano-Villar, S.; Díaz-de Santiago, A.; Sastre, B.; Gutiérrez, C.; Dronda, F.; Coronel Díaz, M.; Domínguez, E.; López-Huertas, M.R.; et al. Maraviroc Is Associated with Latent HIV-1 Reactivation through NF-κB Activation in Resting CD4. J. Virol. 2018, 92, e01931–e02017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Huertas, M.R.; Jiménez-Tormo, L.; Madrid-Elena, N.; Gutiérrez, C.; Rodríguez-Mora, S.; Coiras, M.; Alcamí, J.; Moreno, S. The CCR5-antagonist Maraviroc reverses HIV-1 latency in vitro alone or in combination with the PKC-agonist Bryostatin-1. Sci. Rep. 2017, 7, 2385. [Google Scholar] [CrossRef] [PubMed]
- Cillo, A.R.; Hilldorfer, B.B.; Lalama, C.M.; McKinnon, J.E.; Coombs, R.W.; Tenorio, A.R.; Fox, L.; Gandhi, R.T.; Ribaudo, H.; Currier, J.S.; et al. Virologic and immunologic effects of adding maraviroc to suppressive antiretroviral therapy in individuals with suboptimal CD4+ T-cell recovery. AIDS 2015, 29, 2121–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertas, M.C.; Massanella, M.; Llibre, J.M.; Ballestero, M.; Buzon, M.J.; Ouchi, D.; Esteve, A.; Boix, J.; Manzardo, C.; Miró, J.M.; et al. Intensification of a raltegravir-based regimen with maraviroc in early HIV-1 infection. AIDS 2014, 28, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Geretti, A.M.; Smith, C.; Haberl, A.; Garcia-Diaz, A.; Nebbia, G.; Johnson, M.; Phillips, A.; Staszewski, S. Determinants of virological failure after successful viral load suppression in first-line highly active antiretroviral therapy. Antivir. Ther. 2008, 13, 927–936. [Google Scholar]
- Nicastri, E.; Palmisano, L.; Sarmati, L.; D’Ettorre, G.; Parisi, S.; Andreotti, M.; Buonomini, A.; Pirillo, F.M.; Narciso, P.; Bellagamba, R.; et al. HIV-1 residual viremia and proviral DNA in patients with suppressed plasma viral load (<400 HIV-RNA cp/mL) during different antiretroviral regimens. Curr. HIV Res. 2008, 6, 261–266. [Google Scholar] [CrossRef]
- Riddler, S.A.; Haubrich, R.; DiRienzo, A.G.; Peeples, L.; Powderly, W.G.; Klingman, K.L.; Garren, K.W.; George, T.; Rooney, J.F.; Brizz, B.; et al. Class-sparing regimens for initial treatment of HIV-1 infection. N. Engl. J. Med. 2008, 358, 2095–2106. [Google Scholar] [CrossRef] [Green Version]
- Bonora, S.; Nicastri, E.; Calcagno, A.; Gonzalez de Requena, D.; D’Ettorre, G.; Sarmati, L.; Palmisano, L.; Vullo, V.; Di Perri, G.; Andreoni, M. Ultrasensitive assessment of residual HIV viraemia in HAART-treated patients with persistently undetectable plasma HIV-RNA: A cross-sectional evaluation. J. Med. Virol. 2009, 81, 400–405. [Google Scholar] [CrossRef]
- Pozniak, A.; Gupta, R.K.; Pillay, D.; Arribas, J.; Hill, A. Causes and consequences of incomplete HIV RNA suppression in clinical trials. HIV Clin. Trials 2009, 10, 289–298. [Google Scholar] [CrossRef]
- Charpentier, C.; Landman, R.; Laouénan, C.; Joly, V.; Hamet, G.; Damond, F.; Brun-Vézinet, F.; Mentré, F.; Descamps, D.; Yeni, P. Persistent low-level HIV-1 RNA between 20 and 50 copies/mL in antiretroviral-treated patients: Associated factors and virological outcome. J. Antimicrob. Chemother. 2012, 67, 2231–2235. [Google Scholar] [CrossRef]
- Gianotti, N.; Galli, L.; Racca, S.; Salpietro, S.; Cossarini, F.; Spagnuolo, V.; Barda, B.; Canducci, F.; Clementi, M.; Lazzarin, A.; et al. Residual viraemia does not influence 1 year virological rebound in HIV-infected patients with HIV RNA persistently below 50 copies/mL. J. Antimicrob. Chemother. 2012, 67, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggiolo, F.; Callegaro, A.; Cologni, G.; Bernardini, C.; Velenti, D.; Gregis, G.; Quinzan, G.; Soavi, L.; Iannotti, N.; Malfatto, E.; et al. Ultrasensitive assessment of residual low-level HIV viremia in HAART-treated patients and risk of virological failure. J. Acquir. Immune Defic. Syndr. 2012, 60, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Martin-Blondel, G.; Sauné, K.; Vu Hai, V.; Marchou, B.; Delobel, P.; Izopet, J.; Cuzin, L.; Massip, P. Factors associated with a strictly undetectable viral load in HIV-1-infected patients. HIV Med. 2012, 13, 568–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, S.G.; Andreis, S.; Mengoli, C.; Scaggiante, R.; Ferretto, R.; Manfrin, V.; Cruciani, M.; Giobbia, M.; Boldrin, C.; Basso, M.; et al. Baseline cellular HIV DNA load predicts HIV DNA decline and residual HIV plasma levels during effective antiretroviral therapy. J. Clin. Microbiol. 2012, 50, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarmati, L.; Parisi, S.G.; Montano, M.; Andreis, S.; Scaggiante, R.; Galgani, A.; Viscione, M.; Maffongelli, G.; Ricciardi, A.; Andreoni, C.; et al. Nevirapine use, prolonged antiretroviral therapy and high CD4 nadir values are strongly correlated with undetectable HIV-DNA and -RNA levels and CD4 cell gain. J. Antimicrob. Chemother. 2012, 67, 2932–2938. [Google Scholar] [CrossRef]
- Zheng, L.; Bosch, R.J.; Chan, E.S.; Read, S.; Kearney, M.; Margolis, D.M.; Mellors, J.W.; Eron, J.J.; Gandhi, R.T.; the AIDS Clinical Trials Group (ACTG) A5244 Team. Predictors of residual viraemia in patients on long-term suppressive antiretroviral therapy. Antivir. Ther. 2013, 18, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Charpentier, C.; Choquet, M.; Joly, V.; Yeni, P.; Visseaux, B.; Caseris, M.; Brun-Vézinet, F.; Yazdanpanah, Y.; Peytavin, G.; Descamps, D.; et al. Virological outcome at week 48 of three recommended first-line regimens using ultrasensitive viral load and plasma drug assay. J. Antimicrob. Chemother. 2014, 69, 2819–2825. [Google Scholar] [CrossRef] [Green Version]
- Vancoillie, L.; Demecheleer, E.; Callens, S.; Vogelaers, D.; Vandekerckhove, L.; Mortier, V.; Verhofstede, C. Markers associated with persisting low-level viraemia under antiretroviral therapy in HIV-1 infection. J. Antimicrob. Chemother. 2014, 69, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Kiselinova, M.; Geretti, A.M.; Malatinkova, E.; Vervisch, K.; Beloukas, A.; Messiaen, P.; Bonczkowski, P.; Trypsteen, W.; Callens, S.; Verhofstede, C.; et al. HIV-1 RNA and HIV-1 DNA persistence during suppressive ART with PI-based or nevirapine-based regimens. J. Antimicrob. Chemother. 2015, 70, 3311–3316. [Google Scholar] [CrossRef] [Green Version]
- Konstantopoulos, C.; Ribaudo, H.; Ragland, K.; Bangsberg, D.R.; Li, J.Z. Antiretroviral regimen and suboptimal medication adherence are associated with low-level human immunodeficiency virus viremia. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2015; Volume 2. [Google Scholar] [CrossRef] [Green Version]
- Leierer, G.; Grabmeier-Pfistershammer, K.; Steuer, A.; Geit, M.; Sarcletti, M.; Haas, B.; Kanatschnig, M.; Rappold, M.; Zangerle, R.; Ledergerber, B.; et al. Factors Associated with Low-Level Viraemia and Virological Failure: Results from the Austrian HIV Cohort Study. PLOS ONE 2015, 10, e0142923. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, E.; Castley, A.; Payne, L.; Pummer, S.; Nolan, D. Determinants of residual viraemia during combination HIV treatment: Impacts of baseline HIV RNA levels and treatment choice. HIV Med. 2016, 17, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddler, S.A.; Aga, E.; Bosch, R.J.; Bastow, B.; Bedison, M.; Vagratian, D.; Vaida, F.; Eron, J.J.; Gandhi, R.T.; Mellors, J.W.; et al. Continued Slow Decay of the Residual Plasma Viremia Level in HIV-1-Infected Adults Receiving Long-term Antiretroviral Therapy. J. Infect. Dis. 2016, 213, 556–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianotti, N.; Galli, L.; Galizzi, N.; Ripa, M.; Andolina, A.; Nozza, S.; Spagnuolo, V.; Poli, A.; Lazzarin, A.; Castagna, A. Time spent with residual viraemia after virological suppression below 50 HIV-RNA copies/mL according to type of first-line antiretroviral regimen. Int. J. Antimicrob. Agents 2018, 52, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Geretti, A.M.; White, E.; Orkin, C.; Tostevin, A.; Tilston, P.; Chadwick, D.; Leen, C.; Sabin, C.; Dunn, D.T.; UK HIV Drug Resistance Database; et al. Virological outcomes of boosted protease inhibitor-based first-line ART in subjects harbouring thymidine analogue-associated mutations as the sole form of transmitted drug resistance. J. Antimicrob. Chemother. 2019, 74, 746–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert-Niclot, S.; Boyd, A.; Fofana, D.; Valin, N.; Wirden, M.; Meynard, J.L.; Palich, R.; Agher, R.; Valantin, M.A.; Calvez, V.; et al. INSTI-Based Triple Regimens in Treatment-Naïve HIV-Infected Patients Are Associated With HIV-RNA Viral Load Suppression at Ultralow Levels. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2019; Volume 6. [Google Scholar] [CrossRef] [Green Version]
- Darcis, G.; Maes, N.; Pasternak, A.O.; Sauvage, A.S.; Frippiat, F.; Meuris, C.; Uurlings, F.; Lecomte, M.; Léonard, P.; Elmoussaoui, M.; et al. Detectability of HIV Residual Viremia despite Therapy Is Highly Associated with Treatment with a Protease Inhibitor-Based Combination Antiretroviral Therapy. Antimicrob. Agents Chemother. 2020, 64, e01902-19. [Google Scholar] [CrossRef]
- Jilek, B.L.; Zarr, M.; Sampah, M.E.; Rabi, S.A.; Bullen, C.K.; Lai, J.; Shen, L.; Siliciano, R.F. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat. Med. 2012, 18, 446–451. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Peterson, S.; Sedaghat, A.R.; McMahon, M.A.; Callender, M.; Zhang, H.; Zhou, Y.; Pitt, E.; Anderson, K.S.; Acosta, E.P.; et al. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 2008, 14, 762–766. [Google Scholar] [CrossRef] [Green Version]
- Rabi, S.A.; Laird, G.M.; Durand, C.M.; Laskey, S.; Shan, L.; Bailey, J.R.; Chioma, S.; Moore, R.D.; Siliciano, R.F. Multi-step inhibition explains HIV-1 protease inhibitor pharmacodynamics and resistance. J. Clin. Investig. 2013, 123, 3848–3860. [Google Scholar] [CrossRef]
- Yeh, R.F.; Rezk, N.L.; Kashuba, A.D.; Dumond, J.B.; Tappouni, H.L.; Tien, H.C.; Chen, Y.C.; Vourvahis, M.; Horton, A.L.; Fiscus, S.A.; et al. Genital tract, cord blood, and amniotic fluid exposures of seven antiretroviral drugs during and after pregnancy in human immunodeficiency virus type 1-infected women. Antimicrob. Agents Chemother. 2009, 53, 2367–2374. [Google Scholar] [CrossRef] [Green Version]
- Maggiolo, F.; Ravasio, L.; Ripamonti, D.; Gregis, G.; Quinzan, G.; Arici, C.; Airoldi, M.; Suter, F. Similar adherence rates favor different virologic outcomes for patients treated with nonnucleoside analogues or protease inhibitors. Clin. Infect. Dis. 2005, 40, 158–163. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, J.L.; Gardner, E.M.; Mannheimer, S.B.; Lifson, A.R.; Esser, S.; Telzak, E.E.; Phillips, A.N. Factors associated with adherence amongst 5295 people receiving antiretroviral therapy as part of an international trial. J. Infect. Dis. 2013, 208, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasternak, A.O.; de Bruin, M.; Bakker, M.; Berkhout, B.; Prins, J.M. High Current CD4+ T Cell Count Predicts Suboptimal Adherence to Antiretroviral Therapy. PLoS ONE 2015, 10, e0140791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiser, S.D.; Guzman, D.; Riley, E.D.; Clark, R.; Bangsberg, D.R. Higher rates of viral suppression with nonnucleoside reverse transcriptase inhibitors compared to single protease inhibitors are not explained by better adherence. HIV Clin. Trials 2004, 5, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, A.O.; Kootstra, N.; Vroom, J.; Wit, F.; de Bruin, M.; de Francesco, D.; Sabin, C.; Winston, A.; Prins, J.; Reiss, P.; et al. Non-nucleoside reverse transcriptase inhibitor-based combination antiretroviral therapy is associated with lower cell-associated HIV RNA and DNA levels as compared with therapy based on protease inhibitors. In Proceedings of the 22nd International AIDS Conference, Amsterdam, Netherlands, 23–27 July 2018. [Google Scholar]
- Trinité, B.; Zhang, H.; Levy, D.N. NNRTI-induced HIV-1 protease-mediated cytotoxicity induces rapid death of CD4 T cells during productive infection and latency reversal. Retrovirology 2019, 16, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, A.; Moore, K.L.; Mak, J.; Sluis-Cremer, N.; de Bethune, M.P.; Tachedjian, G. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLOS Pathog. 2006, 2, e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochmans, D.; Anders, M.; Keuleers, I.; Smeulders, L.; Kräusslich, H.G.; Kraus, G.; Müller, B. Selective killing of human immunodeficiency virus infected cells by non-nucleoside reverse transcriptase inhibitor-induced activation of HIV protease. Retrovirology 2010, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Zerbato, J.M.; Tachedjian, G.; Sluis-Cremer, N. Nonnucleoside Reverse Transcriptase Inhibitors Reduce HIV-1 Production from Latently Infected Resting CD4. Antimicrob. Agents Chemother. 2017, 61, e01736–e01816. [Google Scholar] [CrossRef] [Green Version]
- Grennan, J.T.; Loutfy, M.R.; Su, D.; Harrigan, P.R.; Cooper, C.; Klein, M.; Machouf, N.; Montaner, J.S.; Rourke, S.; Tsoukas, C.; et al. Magnitude of virologic blips is associated with a higher risk for virologic rebound in HIV-infected individuals: A recurrent events analysis. J. Infect. Dis. 2012, 205, 1230–1238. [Google Scholar] [CrossRef]
- Sungkanuparph, S.; Overton, E.T.; Seyfried, W.; Groger, R.K.; Fraser, V.J.; Powderly, W.G. Intermittent episodes of detectable HIV viremia in patients receiving nonnucleoside reverse-transcriptase inhibitor-based or protease inhibitor-based highly active antiretroviral therapy regimens are equivalent in incidence and prognosis. Clin. Infect. Dis. 2005, 41, 1326–1332. [Google Scholar] [CrossRef]
- Pascom, A.R.; Pinho, R.E.; Rick, F.; Veras, N.M.; Perini, F.B.; Meireles, M.V.; Pereira, G.F.; Benzaken, A.S.; Avelino-Silva, V.I. Comparison of cumulative viraemia following treatment initiation with different antiretroviral regimens: A real-life study in Brazil. J. Int. AIDS Soc. 2019, 22, e25397. [Google Scholar] [CrossRef]
- Morón-López, S.; Navarro, J.; Jimenez, M.; Rutsaert, S.; Urrea, V.; Puertas, M.C.; Torrella, A.; De Clercq, L.; Ribas, B.P.; Gálvez, C.; et al. Switching From a Protease Inhibitor-based Regimen to a Dolutegravir-based Regimen: A Randomized Clinical Trial to Determine the Effect on Peripheral Blood and Ileum Biopsies From Antiretroviral Therapy-suppressed Human Immunodeficiency Virus-infected Individuals. Clin. Infect. Dis. 2019, 69, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Haïm-Boukobza, S.; Morand-Joubert, L.; Flandre, P.; Valin, N.; Fourati, S.; Sayon, S.; Lavignon, M.; Simon, A.; Girard, P.M.; Katlama, C.; et al. Higher efficacy of nevirapine than efavirenz to achieve HIV-1 plasma viral load below 1 copy/mL. AIDS 2011, 25, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Antinori, A.; Perno, C.F.; Giancola, M.L.; Forbici, F.; Ippolito, G.; Hoetelmans, R.M.; Piscitelli, S.C. Efficacy of cerebrospinal fluid (CSF)-penetrating antiretroviral drugs against HIV in the neurological compartment: Different patterns of phenotypic resistance in CSF and plasma. Clin. Infect. Dis. 2005, 41, 1787–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune activation and HIV persistence: Implications for curative approaches to HIV infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef] [Green Version]
- Hileman, C.O.; Funderburg, N.T. Inflammation, Immune Activation, and Antiretroviral Therapy in HIV. Curr. HIV AIDS Rep. 2017, 14, 93–100. [Google Scholar] [CrossRef]
- Hileman, C.O.; Kinley, B.; Scharen-Guivel, V.; Melbourne, K.; Szwarcberg, J.; Robinson, J.; Lederman, M.M.; McComsey, G.A. Differential Reduction in Monocyte Activation and Vascular Inflammation With Integrase Inhibitor-Based Initial Antiretroviral Therapy Among HIV-Infected Individuals. J. Infect. Dis. 2015, 212, 345–354. [Google Scholar] [CrossRef]
- Kelesidis, T.; Tran, T.T.; Stein, J.H.; Brown, T.T.; Moser, C.; Ribaudo, H.J.; Dube, M.P.; Murphy, R.; Yang, O.O.; Currier, J.S.; et al. Changes in Inflammation and Immune Activation With Atazanavir-, Raltegravir-, Darunavir-Based Initial Antiviral Therapy: ACTG 5260s. Clin. Infect. Dis. 2015, 61, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Kelesidis, T.; Moser, C.; Stein, J.H.; Brown, T.T.; Tran, T.T.; Ribaudo, H.J.; Dube, M.P.; Yang, O.O.; Currier, J.S.; McComsey, G.A. Changes in Markers of T-Cell Senescence and Exhaustion With Atazanavir-, Raltegravir-, and Darunavir-Based Initial Antiviral Therapy: ACTG 5260s. J. Infect. Dis. 2016, 214, 748–752. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Z.; Etemad, B.; Ahmed, H.; Aga, E.; Bosch, R.J.; Mellors, J.W.; Kuritzkes, D.R.; Lederman, M.M.; Para, M.; Gandhi, R.T. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 2016, 30, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, A.O.; Grijsen, M.L.; Wit, F.W.; Bakker, M.; Jurriaans, S.; Prins, J.M.; Berkhout, B. Cell-associated HIV-1 RNA predicts viral rebound and disease progression after discontinuation of temporary early ART. JCI Insight 2020, 5, e134196. [Google Scholar] [CrossRef]
- Moreno, S.; Perno, C.F.; Mallon, P.W.; Behrens, G.; Corbeau, P.; Routy, J.P.; Darcis, G. Two-drug vs. three-drug combinations for HIV-1: Do we have enough data to make the switch? HIV Med. 2019, 20 (Suppl. 4), 2–12. [Google Scholar] [CrossRef] [Green Version]
- Eron, J.; Hung, C.C.; Baril, J.G.; Slim, J.; Falcó, V.; Bogner, J.; Maggiolo, F.; Mills, A.; Sievers, J.; Man, C.Y.; et al. Virologic Response by Baseline Viral Load With Dolutegravir Plus Lamivudine vs Dolutegravir Plus Tenofovir Disoproxil Fumarate/Emtricitabine: Pooled Analysis. J. Acquir. Immune Defic. Syndr. 2020, 84, 60–65. [Google Scholar] [CrossRef]


{kind=link}
| Study (in Chronological Order) | Design | n Total | n by ART Regimens | ART Regimens Compared | Outcome Measure | Difference NNRTI vs. PI | Ratio Univariable (95% CI) | p Uni | Ratio Multivariable (95% CI) | p Multi |
|---|---|---|---|---|---|---|---|---|---|---|
| Palmisano 2005 [51] | Cross-sectional | 84 | 56 (NNRTI), 22 (PI), 6 (NRTI) | Current NNRTI vs. PI | % with RV (> 2.5 copies/mL) | NNRTI < PI | 0.048 | aORiNNRTI = 0.32 (0.12−0.86) | 0.02 | |
| Initial NNRTI vs. PI | NNRTI < PI | 0.009 | aORNNRTI = 0.31 (0.13−0.77) | 0.01 | ||||||
| Maldarelli 2007 [69] | Cross-sectional | 158 | 130 (PI), 28 (NNRTI) | Current NNRTI vs. PI | RV (SCAii) | No difference | 0.29 | |||
| Geretti 2008 [117] | Longitudinal | 1386 | 539 (NNRTI), 457 (PI), 125 (NRTI), 165 (other) | ART regimens when first achieving pVLiii < 50 copies/mL | % with low-level VL rebound (50–400 copies/mL) | NNRTI < PI | ORivbPI = 1.37 (1.00–1.90), ORPI = 1.70 (1.19–2.43) | 0.0005 | aORbPI = 1.39 (1.00−1.94), aORPI = 1.48 (0.97−2.26) | 0.01 |
| % with virological failure (confirmed pVL > 400 copies/mL) | NNRTI < PI (trend) | ORbPI = 1.98 (1.08–3.62), ORPI = 1.28 (0.69–2.38) | 0.06 | aORbPI = 1.88 (1.02−3.46), aORPI = 1.23 (0.66−2.31) | 0.09 | |||||
| Nicastri 2008 [118] | Cross-sectional | 319 | 104 (PI), 166 (NNRTI), 49 (NRTI) | Current ART regimens | RV (LoDv = 2.5 copies/mL) | No difference | 0.5 | |||
| % with RV above the median | NNRTI < PI | 0.002 | ||||||||
| Riddler 2008 [119] | Randomized trial, Longitudinal | 753 | 250 (EFVvi-based), 253 (LPV/rvii-based), 250 (NRTI-sparing) | ART regimens | time to virological failure | EFV < LPV/r | HRviiiEFV = 0.63 (0.45–0.87) | 0.006 | ||
| % with pVL < 50 copies/mL at 96 weeks ART | EFV < LPV/r | 0.003 | ||||||||
| Bonora 2009 [120] | Cross-sectional | 154 | 48 (NVPix), 57 (EFV), 49 (LPV/r) | Current ART regimens | % with RV ( > 2.5 copies/mL) | NVP < other drugs | aORNVP = 0.19−0.82 | 0.013 | ||
| LPV/r > other drugs | aORLPV/r = 0.91−5.17 | 0.08 | ||||||||
| Pozniak 2009 [121] | Systematic review of 15 randomized trials | 8083 | 4475 (NNRTI), 3608 (PI) | First-line NNRTI vs. first-line PI | % with pVL between 50–400 copies/mL at 48 weeks ART | NNRTI < PI | < 0.001 | |||
| Charpentier 2012 [122] | Longitudinal | 656 | 321 (PI), 220 (NNRTI), 115 (other) | ART regimens | % with low-level viremia (20–50 copies/mL) | No difference | 0.23 | |||
| Doyle 2012 [47] | Longitudinal | 1247 | 268 (NNRTI), 492 (PI), 30 (NRTI), 101 (other) | ART regimens | % with pVL between 40–49 copies/mL vs. detectable < 40 copies/mL vs. undetectable | NNRTI < PI | < 0.0001 | |||
| % with viral rebound to > 50 copies/mL within one year | NNRTI < PI | HRNNRTI = 0.27 (0.16–0.45) | < 0.0001 | aHRxNNRTI = 0.40 (0.21−0.77) | 0.002 | |||||
| % with viral rebound to >400 copies/mL within one year | NNRTI < PI | HRNNRTI = 0.32 (0.14–0.71) | 0.007 | aHRNNRTI = 0.46 (0.17−1.23) | 0.23 | |||||
| Gianotti 2012 [123] | Longitudinal | 739 | 204 (NNRTI), 414 (PI), 46 (NRTI), 75 (other) | ART regimens | % with RV ( > 1 copy/mL) | NNRTI < PI | 0.001 | |||
| Maggiolo 2012 [124] | Longitudinal | 1214 | 666 (NNRTI), 450 (PI), 98 (other) | Current NNRTI vs. PI | Risk of virological failure | NNRTI < PI | < 0.0001 | |||
| % with RV ( > 3 copies/mL) | NNRTI < PI | < 0.0001 | ||||||||
| Martin-Blondel 2012 [125] | Cross-sectional | 1392 | 45% (NNRTI), 43% (PI), 12% (INSTI) | ART regimens | % with pVL between 20–50 copies/mL vs. detectable < 20 copies/mL vs. undetectable | NNRTI < PI | 0.0008 | aORNNRTI = 1.45 (1.03−2.04) | 0.03 | |
| Parisi 2012 [126] | Cross-sectional | 180 | 71 (EFV), 21 (NVP), 83 (PI), 5 (other) | ART regimens | % with pVL 50–1000 copies/mL vs. 21–49 copies/mL vs. 2.5–20 copies/mL vs. < 2.5 copies/mL | No difference | NSxi | |||
| Sarmati 2012 [127] | Cross-sectional | 420 | 228 (NNRTI), 192 (PI) | Current NNRTI vs. PI | % without RV ( < 1 copy/mL) | NNRTI < PI | ORNNRTI = 1.73 (1.15–2.60) | 0.008 | ||
| Zheng 2013 [128] | Cross-sectional | 103 | 65% (NNRTI), 35% (PI) | Current NNRTI vs. PI | RV (SCA) | No difference | NS | |||
| Charpentier 2014 [129] | Cross-sectional | 168 | 60 (EFV), 108 (PI) | ART regimens | Virological outcome at week 48 ART (pVL < 50, < 20, and < 1 copies/mL) | No difference | NS | |||
| Vancoillie 2014 [130] | Longitudinal | 173 | 49 (NNRTI), 122 (PI), 2 (other) | Initial NNRTI vs. PI | % with long-term low-level viremia (20–250 copies/mL) vs. undetectable pVL | NNRTI < PI | 0.002 | aORPI = 2.90 (1.20–6.97) | 0.017 | |
| Kiselinova 2015 [131] | Case-control | 161 | 81 (NVP), 80 (PI) | Current NVP vs. PI | % without RV (SCA) | No difference | ORNVP = 1.53 (0.82–2.86) | 0.17 | ||
| Konstantopoulos 2015 [132] | Cross-sectional | 128 | 45 (NNRTI), 83 (PI) | Current NNRTI vs. PI | % with low-level viremia (50–1000 copies/mL) | NNRTI < PI | HRbPI = 2.7 (1.1–6.4) | 0.03 | aHRbPIxii = 3.1 (1.3−7.4) | 0.01 |
| HRPI = 3.0 (1.1–6.4) | 0.03 | aHRPI = 3.1 (1.2–8.3) | 0.02 | |||||||
| Leierer 2015 [133] | Cross-sectional | 2276 | 1300 (NNRTI- or INSTI), 976 (PI) | Current NNRTI/INSTI vs. PI | % with low-level viremia ( < 200 copies/mL) vs. BLQxiii | NNRTI/INSTI < PI | ORPI = 1.52 (1.15–2.01) | 0.003 | aORPI = 1.54 (1.15–2.06) | |
| % with virological failure (pVL ≥ 200 copies/mL) vs. BLQ | NNRTI/INSTI < PI | ORPI = 2.78 (1.74–4.42) | < 0.001 | aORPI = 2.36 (1.45–3.83) | ||||||
| McKinnon 2016 [134] | Longitudinal | 356 | 204 (NNRTI), 152 (PI) | Current NNRTI vs. PI | % with pVL < 40 copies/mL among those with pVL < 200 copies/mL | NNRTI < PIxiv | ORNNRTI = 2.26 (1.30–3.94) | 0.004 | aORNNRTI = 2.06 (0.99–4.28) | 0.05 |
| NNRTI < PIxv | ORNNRTI = 1.98 (1.17–3.37) | 0.01 | aORNNRTI = 1.99 (1.14–3.48) | 0.02 | ||||||
| % with RV among those with pVL < 40 copies/mL (n = 348) | No differencexiv | ORNNRTI = 0.77 (0.52–1.14) | 0.2 | aORNNRTI = 0.86 (0.56–1.31) | 0.5 | |||||
| NNRTI < PIxv | ORNNRTI = 0.53 (0.34–0.85) | 0.008 | aORNNRTI = 0.54 (0.34–0.86) | 0.01 | ||||||
| Riddler 2016 [135] | Cross-sectional | 334 | 61% (NNRTI), 28% (PI), 11% (other) | Initial NNRTI vs. PI | % with RV at 192 and 208 weeks of ART (SCA) | No difference | ORPI = 1.16 (0.79–1.61) | 0.45 | aORPI = 1.30 (0.88–1.92) | 0.19 |
| Gianotti 2018 [136] | Longitudinal | 771 | 244 (NNRTI), 254 (PI), 234 (INSTI), 39 (other) | First-line NNRTI vs. first-line PI | % of time on ART spent with RV (detectable < 50 copies/mL) | NNRTI < PI | < 0.0001 | |||
| Geretti 2019 [137] | Longitudinal | 6599 | 4889 (NNRTI), 1710 (PI) | First-line NNRTI vs. first-line PI | % with virological suppression on ART | NNRTI < PI | HRPI = 0.69 | aHRPI = 0.70 (0.65–0.74) | < 0.001 | |
| % with viremia ( > 50 copies/mL) | NNRTI < PI | HRPI = 2.27 | aHRPI = 2.17 (1.88–2.51) | < 0.001 | ||||||
| Lambert-Niclot 2019 [138] | Longitudinal | 717 | 211 (NNRTI), 419 (PI), 87 (INSTI) | First-line ART regimens | % achieving ultralow VL - not detected on ART | No difference | ||||
| % with virological rebound on ART | NNRTI < PI | HRNNRTI = 0.60 (0.43–0.84)xvi | 0.003 | aHRNNRTI = 0.76 (0.50–1.15) | 0.2 | |||||
| HRPI = 1.20 (0.88–1.64)xvi | 0.2 | aHRPI = 1.00 (0.69–1.43) | 0.9 | |||||||
| Darcis 2020 [139] | Longitudinal | 1160 | Samples: 4210 (NNRTI), 3280 (PI), 3555 (INSTI) | Current NNRTI vs. PI | % samples with detectable RV (all samples < 20 copies/mL) | NNRTI < PI | aORNNRTI = 0.85 (0.74–0.97) | 0.013 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darcis, G.; Berkhout, B.; Pasternak, A.O. Differences in HIV Markers between Infected Individuals Treated with Different ART Regimens: Implications for the Persistence of Viral Reservoirs. Viruses 2020, 12, 489. https://doi.org/10.3390/v12050489
Darcis G, Berkhout B, Pasternak AO. Differences in HIV Markers between Infected Individuals Treated with Different ART Regimens: Implications for the Persistence of Viral Reservoirs. Viruses. 2020; 12(5):489. https://doi.org/10.3390/v12050489
Chicago/Turabian StyleDarcis, Gilles, Ben Berkhout, and Alexander O. Pasternak. 2020. "Differences in HIV Markers between Infected Individuals Treated with Different ART Regimens: Implications for the Persistence of Viral Reservoirs" Viruses 12, no. 5: 489. https://doi.org/10.3390/v12050489