
Full-Length Computational Model of the SARS-CoV-2 Spike Protein and Its Implications for a Viral Membrane Fusion Mechanism


Abstract
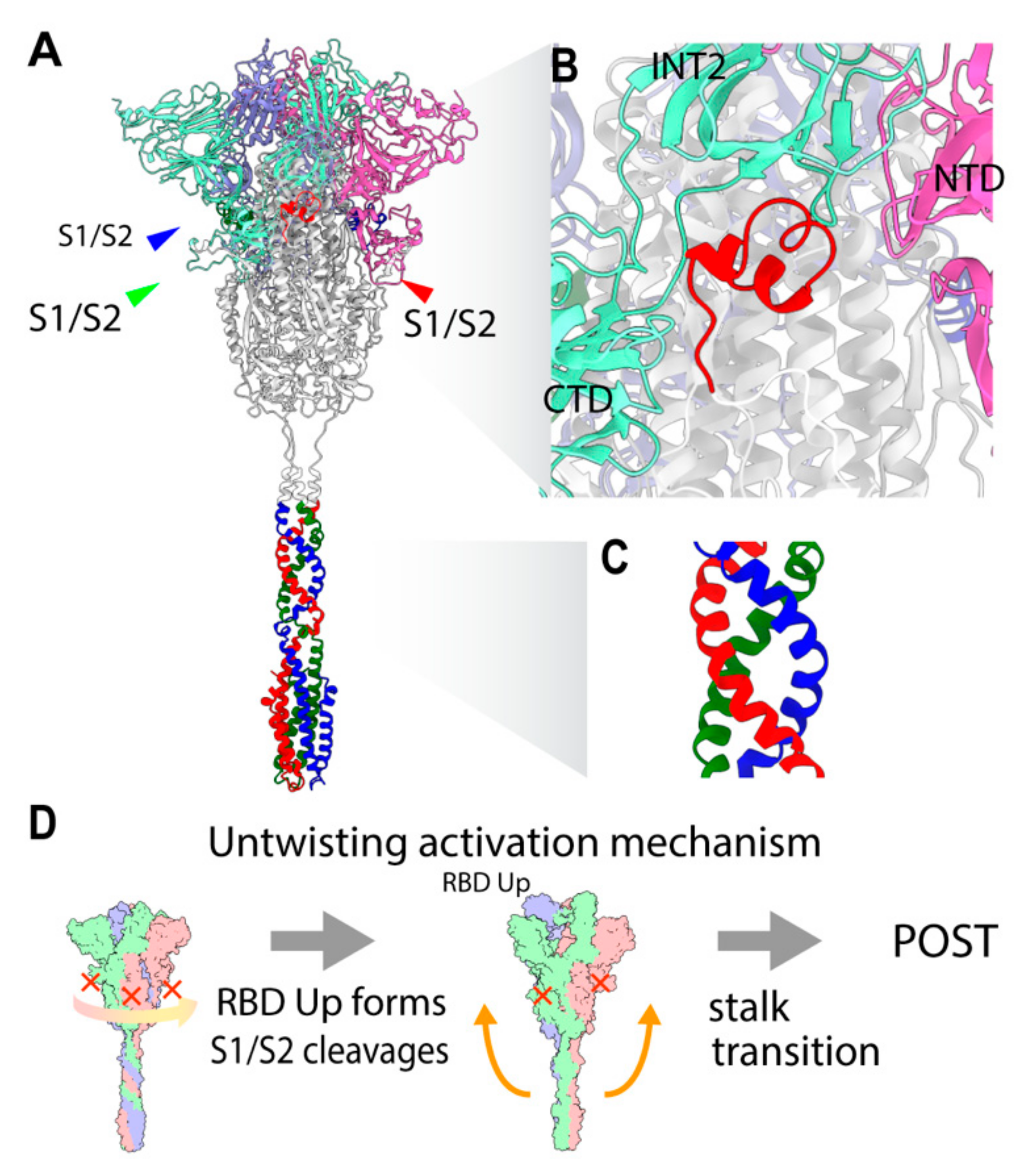
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
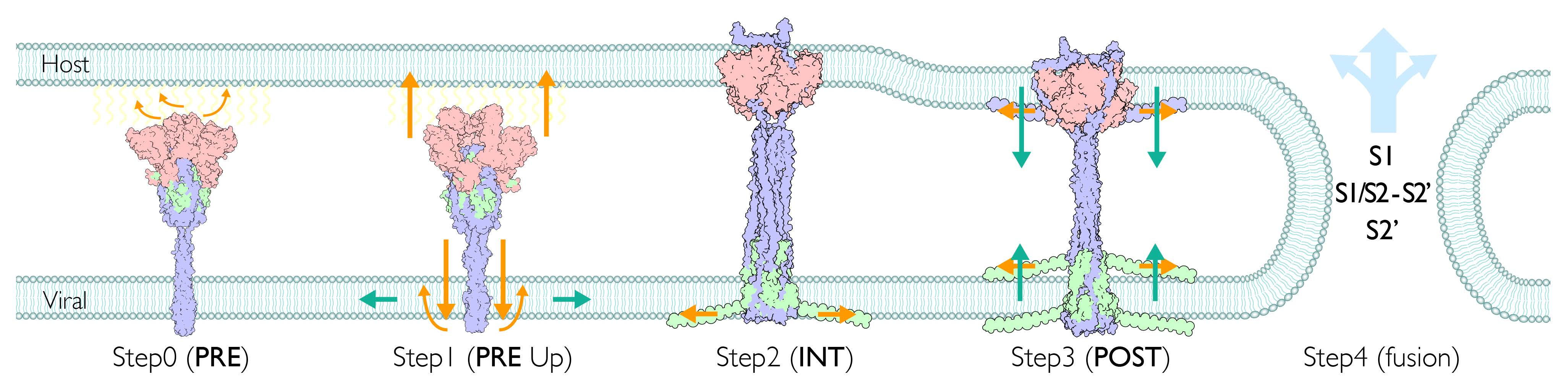
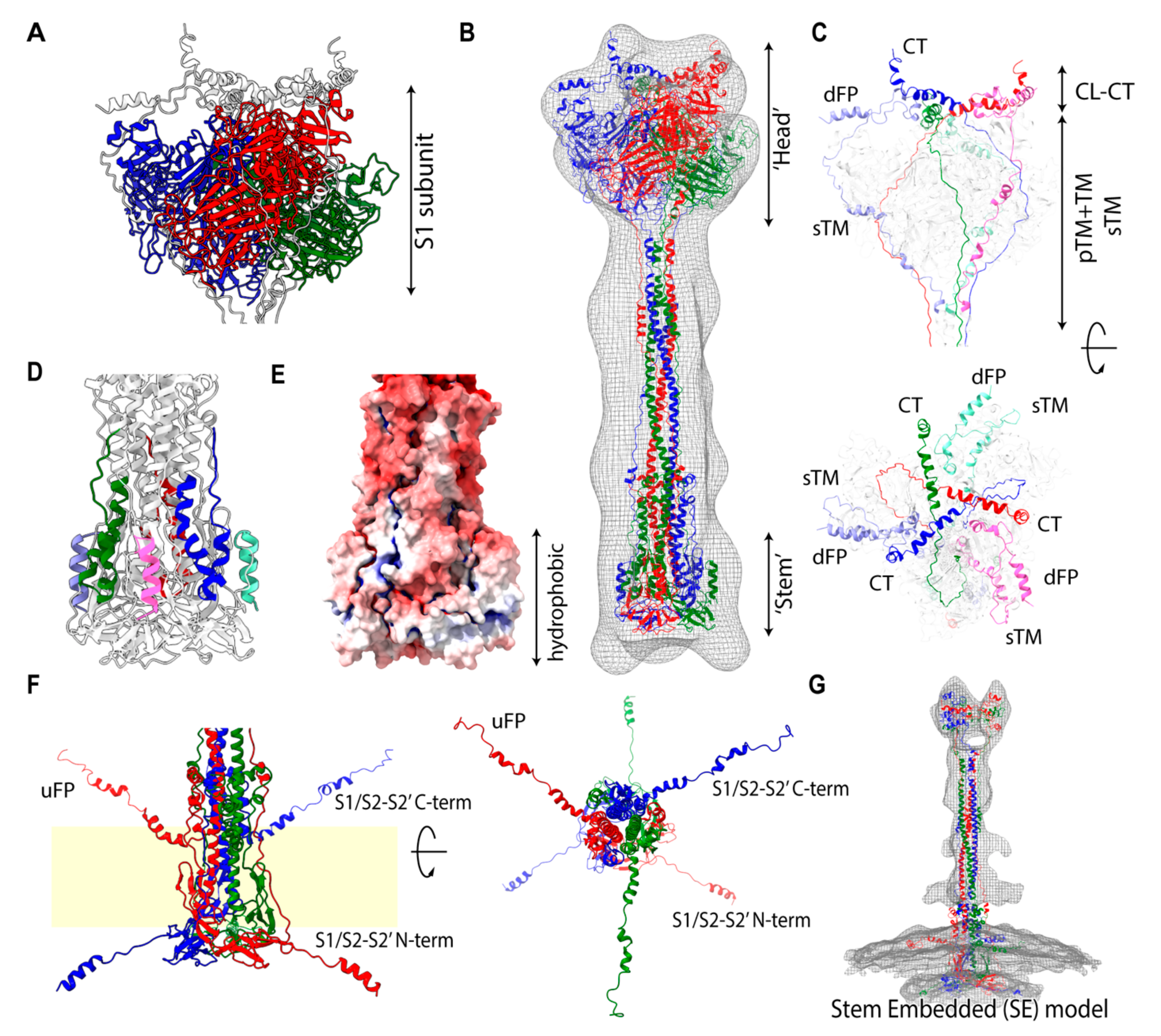
2.1. Assignment of Spike Protein Segments is Functionally Crucial for the Viral Membrane Fusion
2.2. Spike Transmembrane in the PRE State Is Characterized by Left-Handed Coiled-Coil Winding
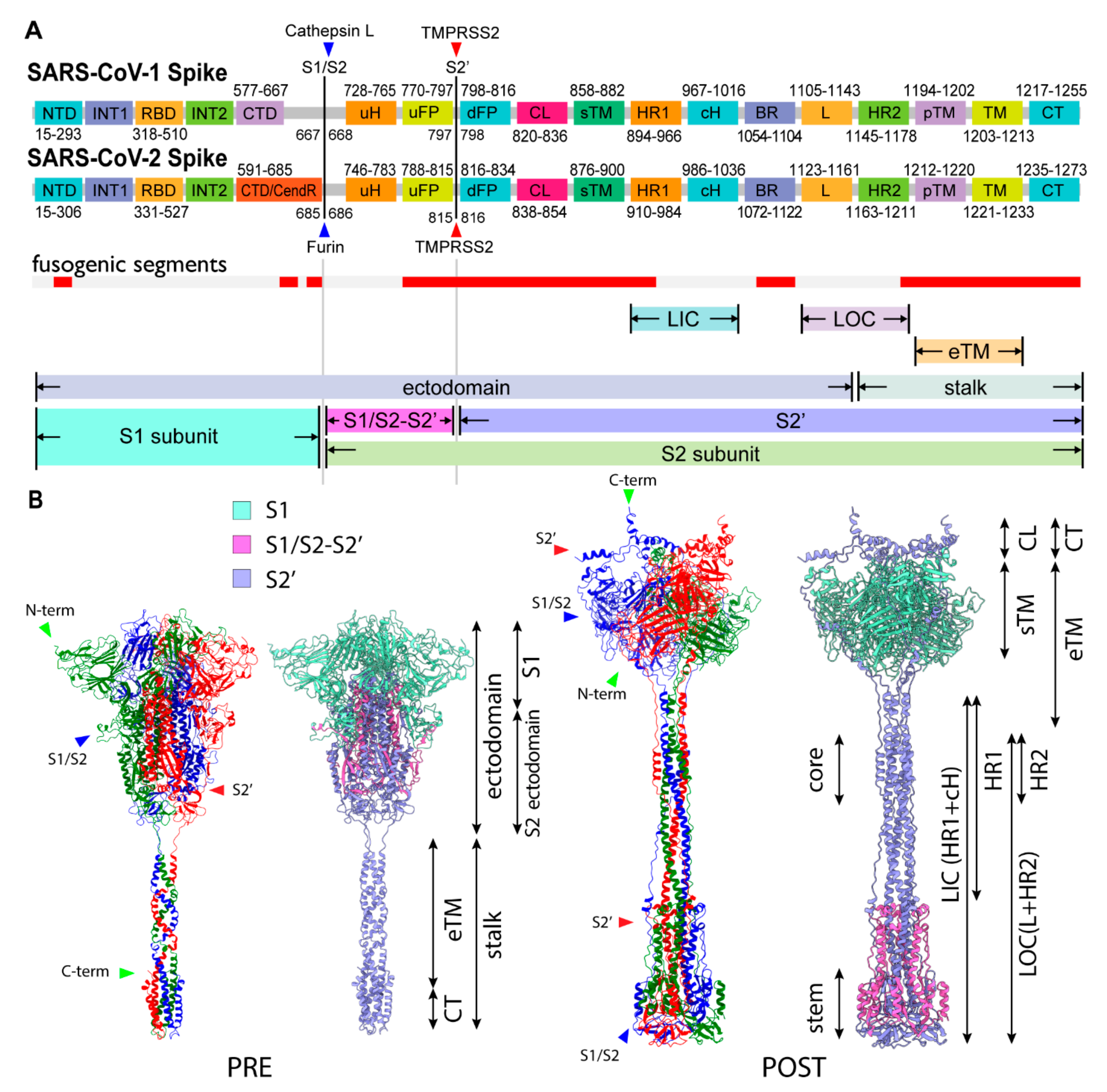
2.3. Three S1/S2 Cleavages and Three Up RBDs Enable S2 Ectodomain to Rotate for the Structural Transition
2.4. S2 is Capable of Maintaining the Attachment with an S1 Trimer in Solution
2.5. Stem-Embedded Model Reveals a New Assignment into the POST-Like Density Map on the Virion
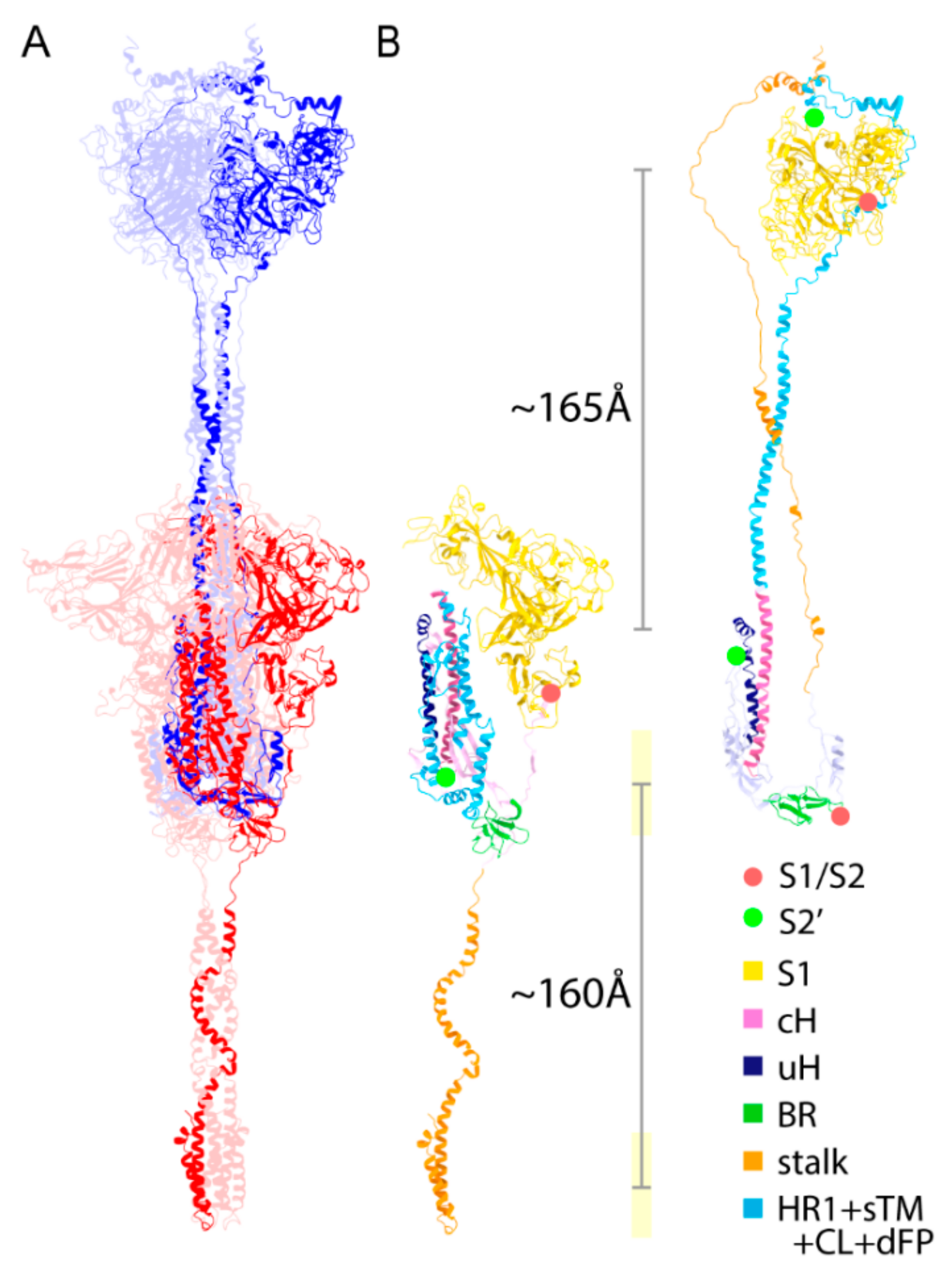
2.6. Structural Comparison for the Transition from the PRE State toward the POST State
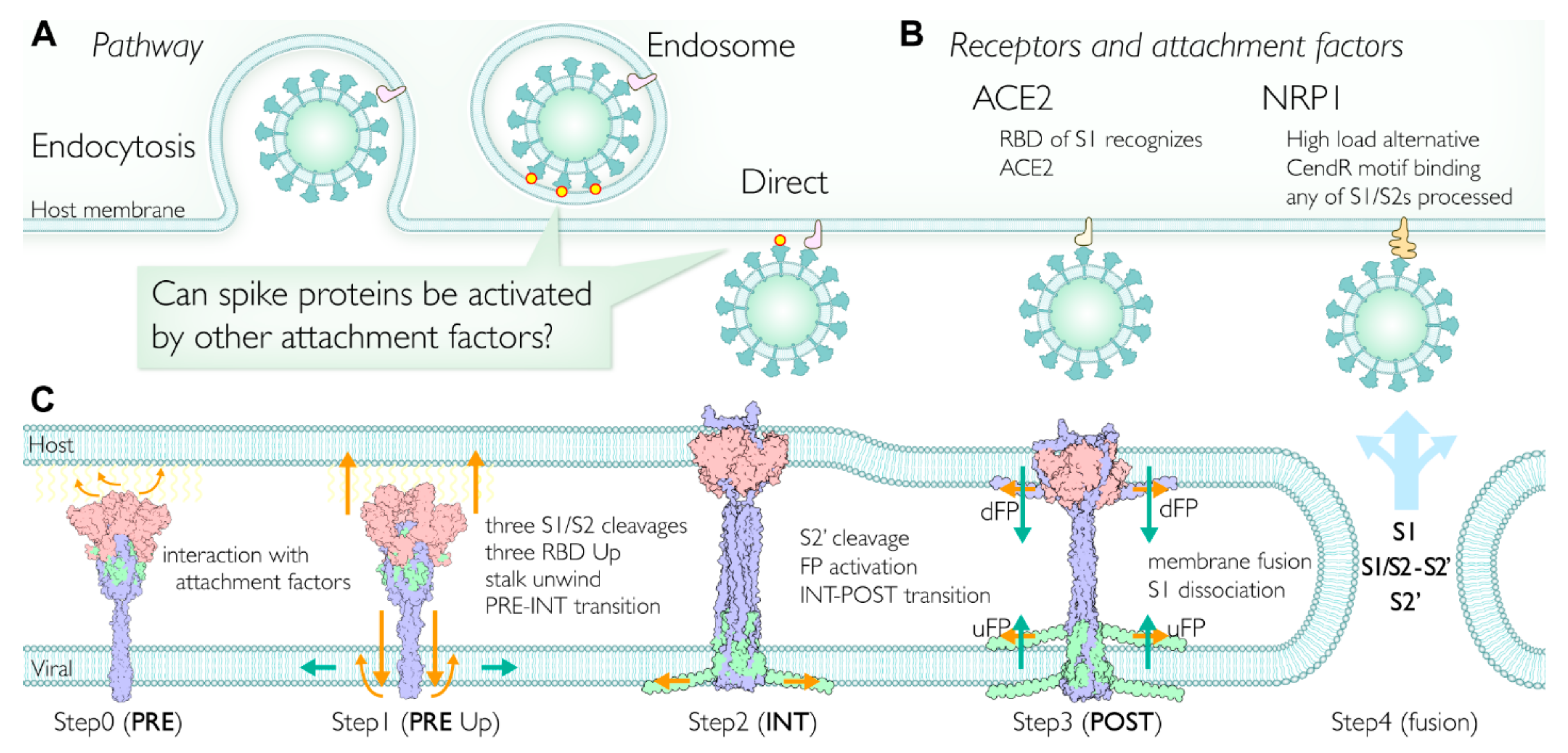
2.7. Multiple Pathways Initiate the Viral Membrane Fusion after Binding to the Receptor
2.8. Involvment of S1/S2 Cleavage and Untwisting Activation Mechanism in the Structural Transition
2.9. Extended Transmembrane (eTM) Wraps around a Trimeric S1 and Subsequently a Long Internal Core (LIC) Forms a Long Coiled-Coil toward the POST State
2.10. Role of Intermediate (INT) State Realized after the Transition before the S2′ Cleavage
2.11. Initiation of Membrane Fusion and S1 Trimer Release
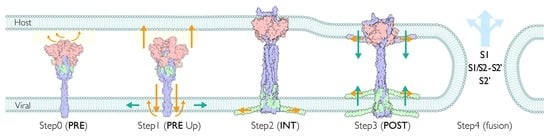
2.12. Viral Membrane Fusion Mechanism: Proteolytic Cleavage Events, Distinct States, and Membrane Fusion
2.13. Comparison with Conventional Steps Mediated by Viral Class I Fusion Protein
2.14. Case Studies
2.15. Case Study 1: Effect of Neutralizing Antibody Binding to the Spike Protein
2.16. Case Study 2: D614G Variant Has High Infectivity and Transmissibility of COVID-19
3. Concluding Remark
4. Materials and Methods
4.1. Modeling of the PRE State
4.2. Modeling of POST State
Modeling Strategy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data and Materials Availability
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Malik, Y.S.; Kumar, N.; Sircar, S.; Kaushik, R.; Bhat, S.; Dhama, K.; Gupta, P.; Goyal, K.; Singh, M.P.; Ghoshal, U.; et al. Coronavirus disease pandemic (Covid-19): Challenges and a global perspective. Pathogens 2020, 9, 519. [Google Scholar] [CrossRef]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef]
- Chen, Q.; Allot, A.; Lu, Z. LitCovid: An open database of COVID-19 literature. Nucleic Acids Res. 2021, 49, D1534–D1540. [Google Scholar] [CrossRef] [PubMed]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 18. [Google Scholar] [CrossRef]
- Hakansson-McReynolds, S.; Jiang, S.; Rong, L.; Caffrey, M. Solution structure of the severe acute respiratory syndrome-coronavirus heptad repeat 2 domain in the prefusion state. J. Biol. Chem. 2006, 281, 11965–11971. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.-Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a pH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains. Cell Host Microbe 2020, 867–879. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, A.; Paray, B.A.; Hussain, A.; Qadir, F.A.; Attar, F.; Aziz, F.M.; Sharifi, M.; Derakhshankhah, H.; Rasti, B.; Mehrabi, M.; et al. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J. Biomol. Struct. Dyn. 2020, 39, 3025–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Mendonça, L.; Yang, Y.; Gao, Y.; Shen, C.; Liu, J.; Ni, T.; Ju, B.; Liu, C.; Tang, X.; et al. The Architecture of Inactivated SARS-CoV-2 with Postfusion Spikes Revealed by CryoEM and CryoET. Structure 2020, 28, 1218–1224. [Google Scholar] [CrossRef]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.T.; Dutch, R.E. Viral membrane fusion and the transmembrane domain. Viruses 2020, 12, 693. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Cao, D.; Kong, L.; Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 2020, 11, 3618. [Google Scholar] [CrossRef]
- Watanabe, R.; Matsuyama, S.; Shirato, K.; Maejima, M.; Fukushi, S.; Morikawa, S.; Taguchi, F. Entry from the Cell Surface of Severe Acute Respiratory Syndrome Coronavirus with Cleaved S Protein as Revealed by Pseudotype Virus Bearing Cleaved S Protein. J. Virol. 2008, 82, 11985–11991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Ni, D.; Lau, K.; Lehmann, F.; Fränkl, A.; Hacker, D.; Pojer, F.; Stahlberg, H. Structural investigation of ACE2 dependent disassembly of the trimeric SARS-CoV-2 Spike glycoprotein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2020, 5575, 97. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef]
- Guo, H.; Huang, M.; Yuan, Q.; Wei, Y.; Gao, Y.; Mao, L.; Gu, L.; Tan, Y.W.; Zhong, Y.; Liu, D.; et al. The important role of lipid raft-mediated attachment in the infection of cultured cells by coronavirus infectious bronchitis virus beaudette strain. PLoS ONE 2017, 12, e0170123. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- McReynolds, S.; Jiang, S.; Guo, Y.; Celigoy, J.; Schar, C.; Rong, L.; Caffrey, M. Characterization of the prefusion and transition states of severe acute respiratory syndrome coronavirus S2-HR2. Biochemistry 2008, 47, 6802–6808. [Google Scholar] [CrossRef] [PubMed]
- McReynolds, S.; Jiang, S.; Rong, L.; Caffrey, M. Dynamics of SARS-coronavirus HR2 domain in the prefusion and transition states. J. Magn. Reson. 2009, 201, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.; Simonetti, B.; Antón-Plágaro, C.; Kavanagh Williamson, M.; Shoemark, D.; Simón-Gracia, L.; Klein, K.; Bauer, M.; Hollandi, R.; Greber, U.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–886. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Wang, N.; Pallesen, J.; Wrapp, D.; Turner, H.L.; Cottrell, C.A.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 2018, 8, 15701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Tomlinson, A.C.A.; Wong, A.H.M.; Zhou, D.; Desforges, M.; Talbot, P.J.; Benlekbir, S.; Rubinstein, J.L.; Rini, J.M. The human coronavirus HCoV-229E S-protein structure and receptor binding. Elife 2019, 8, 1–22. [Google Scholar] [CrossRef]
- Mahajan, M.; Bhattacharjya, S. NMR structures and localization of the potential fusion peptides and the pre-transmembrane region of SARS-CoV: Implications in membrane fusion. Biochim. Biophys. Acta Biomembr. 2015, 1848, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, M.; Chatterjee, D.; Bhuvaneswari, K.; Pillay, S.; Bhattacharjya, S. NMR structure and localization of a large fragment of the SARS-CoV fusion protein: Implications in viral cell fusion. Biochim. Biophys. Acta Biomembr. 2018, 1860, 407–415. [Google Scholar] [CrossRef]
- Lai, A.L.; Millet, J.K.; Daniel, S.; Freed, J.H.; Whittaker, G.R. The SARS-CoV Fusion Peptide Forms an Extended Bipartite Fusion Platform that Perturbs Membrane Order in a Calcium—Dependent Manner. J. Mol. Biol. 2017, 429, 3875–3892. [Google Scholar] [CrossRef]
- Li, F.; Berardi, M.; Li, W.; Farzan, M.; Dormitzer, P.R.; Harrison, S.C. Conformational States of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein Ectodomain. J. Virol. 2006, 80, 6794–6800. [Google Scholar] [CrossRef] [Green Version]
- Asandei, A.; Mereuta, L.; Schiopu, I.; Park, J.; Seo, C.H.; Park, Y.; Luchian, T. Non-Receptor-Mediated Lipid Membrane Permeabilization by the SARS-CoV-2 Spike Protein S1 Subunit. ACS Appl. Mater. Interfaces 2020, 12, 55649–55658. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M. Fusion peptides and the mechanism of viral fusion. Biochim. Biophys. Acta Biomembr. 2003, 1614, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Basso, L.G.M.; Vicente, E.F.; Crusca, E.; Cilli, E.M.; Costa-Filho, A.J. SARS-CoV fusion peptides induce membrane surface ordering and curvature. Sci. Rep. 2016, 6, 37131. [Google Scholar] [CrossRef] [Green Version]
- Letarov, A.V.; Babenko, V.V.; Kulikov, E.E. Free SARS-CoV-2 Spike Protein S1 Particles May Play a Role in the Pathogenesis of COVID-19 Infection. Biochemistry 2020, 86, 257–261. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Uemura, K.; Sato, A.; Toba, S.; Sanaki, T.; Maenaka, K.; Hall, W.W.; Orba, Y.; Sawa, H. SARS-CoV-2 variants with mutations at the S1/S2 cleavage site are generated in vitro during propagation in TMPRSS2-deficient cells. PLoS Pathog. 2021, 17, e1009233. [Google Scholar] [CrossRef]
- Gomes, C.P.; Fernandes, D.E.; Casimiro, F.; da Mata, G.F.; Passos, M.T.; Varela, P.; Mastroianni-Kirsztajn, G.; Pesquero, J.B. Cathepsin L in COVID-19: From Pharmacological Evidences to Genetics. Front. Cell. Infect. Microbiol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Bansal, V.; Feschotte, C. A Single-Cell RNA Expression Map of Human Coronavirus Entry Factors. Cell Rep. 2020, 32, 108175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Feng, F.; Hu, G.; Wang, Y.; Yu, Y.; Zhu, Y.; Xu, W.; Cai, X.; Sun, Z.; Han, W.; et al. The S1/S2 boundary of SARS-CoV-2 spike protein modulates cell entry pathways and transmission. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sougrat, R.; Bartesaghi, A.; Lifson, J.D.; Bennett, A.E.; Bess, J.W.; Zabransky, D.J.; Subramaniam, S. Electron tomography of the contact between T cells and SIV/HIV-1: Implications for viral entry. PLoS Pathog. 2007, 3, e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhaim, M.A.; Mangala Prasad, V.; Garcia, N.K.; Guttman, M.; Lee, K.K. Structural monitoring of a transient intermediate in the hemagglutinin fusion machinery on influenza virions. Sci. Adv. 2020, 6, eaaz8822. [Google Scholar] [CrossRef]
- Corver, J.; Broer, R.; Van Kasteren, P.; Spaan, W. Mutagenesis of the transmembrane domain of the SARS coronavirus spike glycoprotein: Refinement of the requirements for SARS coronavirus cell entry. Virol. J. 2009, 6, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guille’n, J.; Pe´rez-Berna, A.; Moreno, M.R.; Villalaı´n, J. Identification of the Membrane-Active Regions of the Severe Acute Respiratory Syndrome Coronavirus Spike Membrane Glycoprotein Using a 16/18-Mer Peptide Scan: Implications for the Viral Fusion Mechanism. J. Virol. 2005, 79, 1743–1752. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Zhang, X.; Yang, Y.; Hotez, P.J.; Du, L. Neutralizing antibodies for the treatment of COVID-19. Nat. Biomed. Eng. 2020, 4, 1134–1139. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Walls, A.C.; Xiong, X.; Park, Y.J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019, 176, 1026–1039.e15. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the effects of SARS-CoV-2 Spike mutation D614G on transmissibility and pathogenicity. Cell 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. The circulating SARS-CoV-2 spike variant N439K maintains fitness while evading antibody-mediated immunity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chan, K.K.; Tan, T.J.C.; Narayanan, K.K.; Procko, E. An engineered decoy receptor for SARS-CoV-2 broadly binds protein S sequence variants. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Zhang, S.; Wang, Q.; Anang, S.; Wang, J.; Ding, H.; Kappes, J.C.; Sodroski, J. Spike Glycoprotein and Host Cell Determinants of SARS-CoV-2 Entry and Cytopathic Effects. J. Virol. 2020. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishima, W.; Kulik, M. Full-Length Computational Model of the SARS-CoV-2 Spike Protein and Its Implications for a Viral Membrane Fusion Mechanism. Viruses 2021, 13, 1126. https://doi.org/10.3390/v13061126
Nishima W, Kulik M. Full-Length Computational Model of the SARS-CoV-2 Spike Protein and Its Implications for a Viral Membrane Fusion Mechanism. Viruses. 2021; 13(6):1126. https://doi.org/10.3390/v13061126
Chicago/Turabian StyleNishima, Wataru, and Marta Kulik. 2021. "Full-Length Computational Model of the SARS-CoV-2 Spike Protein and Its Implications for a Viral Membrane Fusion Mechanism" Viruses 13, no. 6: 1126. https://doi.org/10.3390/v13061126