
Genomic Characterisation of a Highly Divergent Siadenovirus (Psittacine Siadenovirus F) from the Critically Endangered Orange-Bellied Parrot (Neophema chrysogaster)

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of Sample, Extraction of DNA, and Confirmation of the Presence of PsSiAdV-F DNA
2.2. Library Construction and Sequencing
2.3. Genome Assembly
2.4. Genome Annotation and Bioinformatics
2.5. Comparative Genomics
2.6. Phylogenetic Analyses
2.7. Recombination Analyses
3. Results
3.1. Genome of PsSiAdV-F
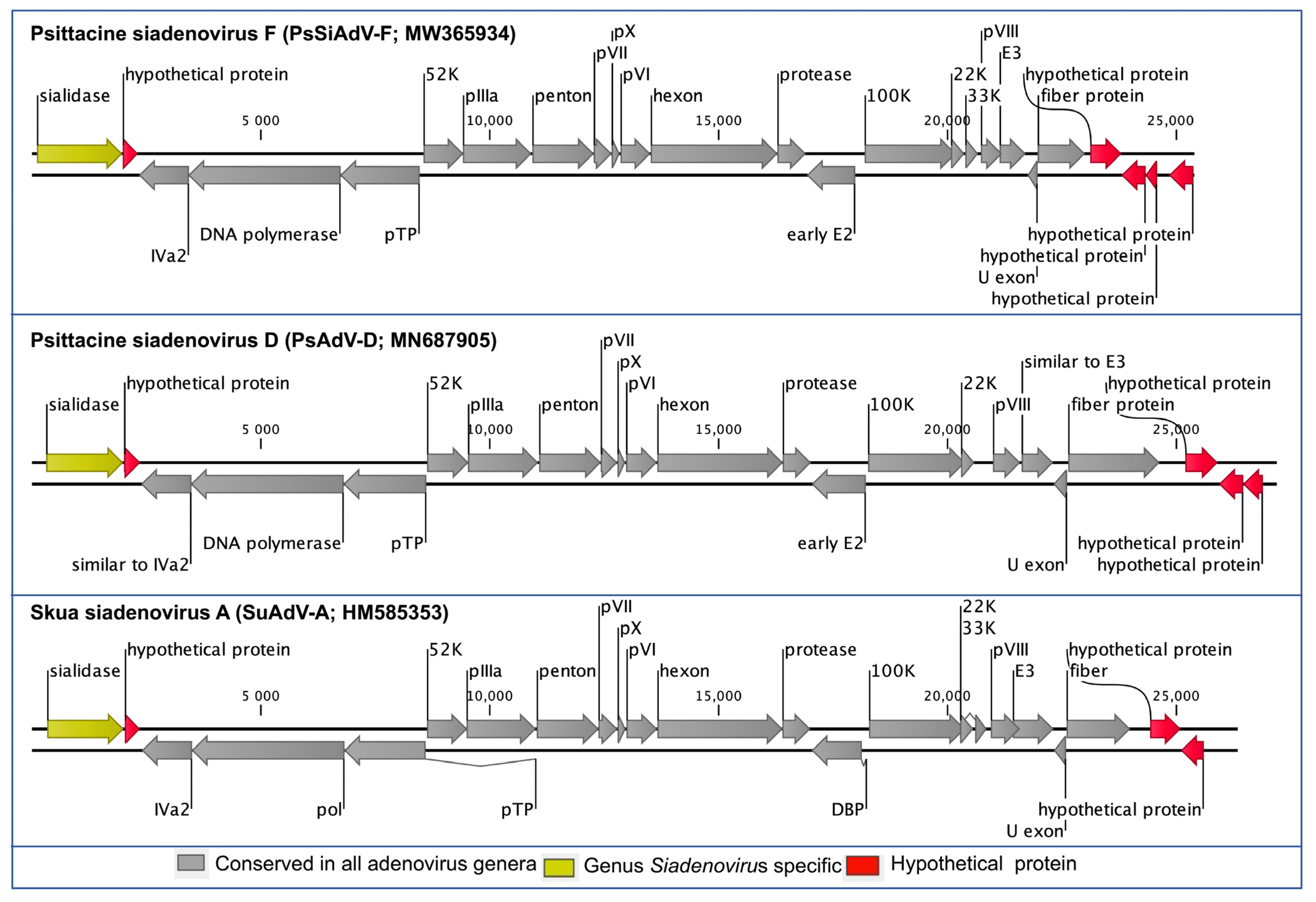
3.2. Genome Annotation and Comparative Analyses of PsSiAdV-F
3.3. Evolutionary Relationships of PsSiAdV-F
3.4. Evidence of a Rare Recombination Event
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harrach, B.; Tarjan, Z.L.; Benko, M. Adenoviruses across the animal kingdom: A walk in the zoo. FEBS Lett. 2019, 593, 3660–3673. [Google Scholar] [CrossRef]
- ICTV. Virus Taxonomy—2020 Release. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 18 August 2021).
- Davison, A.; Wright, K.; Harrach, B. DNA sequence of frog adenovirus. J. Gen. Virol. 2000, 81, 2431–2439. [Google Scholar] [CrossRef]
- Rivera, S.; Wellehan, J.; McManamon, R.; Innis, C.; Garner, M.; Raphael, B.; Gregory, C.; Latimer, K.; Rodriguez, C.; Figueroa, O.; et al. Systemic adenovirus infection in Sulawesi tortoises (Indotestudo forsteni) caused by a novel siadenovirus. J. Vet. Diagn. Invest. 2009, 21, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Vaz, F.F.; Raso, T.F.; Agius, J.E.; Hunt, T.; Leishman, A.; Eden, J.S.; Phalen, D.N. Opportunistic sampling of wild native and invasive birds reveals a rich diversity of adenoviruses in Australia. Virus Evol. 2020, 6, veaa024. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, J.H.; Park, Y.M.; Shin, O.S.; Kim, H.; Choi, H.G.; Song, J.W. A novel adenovirus in Chinstrap penguins (Pygoscelis antarctica) in Antarctica. Viruses 2014, 6, 2052–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.M.; Kim, J.H.; Gu, S.H.; Lee, S.Y.; Lee, M.G.; Kang, Y.K.; Kang, S.H.; Kim, H.J.; Song, J.W. Full genome analysis of a novel adenovirus from the South Polar skua (Catharacta maccormicki) in Antarctica. Virology 2012, 422, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, M.; Sarker, S.; Vaz, P.K.; Legione, A.R.; Devlin, J.M.; Macwhirter, P.L.; Whiteley, P.L.; Raidal, S.R. Disease surveillance in wild Victorian cacatuids reveals co-infection with multiple agents and detection of novel avian viruses. Vet. Microbiol. 2019, 235, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Rinder, M.; Schmitz, A.; Baas, N.; Korbel, R. Molecular identification of novel and genetically diverse adenoviruses in Passeriform birds. Virus Genes 2020, 56, 316–324. [Google Scholar] [CrossRef]
- Zadravec, M.; Racnik, J.; Slavec, B.; Ballmann, M.; Marhold, C.; Harrach, B.; Rojs, O. Detection of New Adenoviruses in Psittacine Birds in Slovenia. In Proceedings of the IX Symposium Poultry Days 2011 with International Participation, Sibenik, Croatia, 11–14 May 2011; Balenovic, C.M., Ed.; Poultry Centre: Zagreb, Croatia, 2011; pp. 36–39. [Google Scholar]
- Wellehan, J.F., Jr.; Greenacre, C.B.; Fleming, G.J.; Stetter, M.D.; Childress, A.L.; Terrell, S.P. Siadenovirus infection in two psittacine bird species. Avian Pathol. 2009, 38, 413–417. [Google Scholar] [CrossRef]
- Phalen, D.N.; Agius, J.; Vaz, F.F.; Eden, J.S.; Setyo, L.C.; Donahoe, S. A survey of a mixed species aviary provides new insights into the pathogenicity, diversity, evolution, host range, and distribution of psittacine and passerine adenoviruses. Avian Pathol. 2019, 48, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; McLelland, J.; McLelland, D.J.; Clarke, J.; Woolford, L.; Eden, P.; Phalen, D.N. Psittacid Adenovirus-2 infection in the critically endangered orange-bellied parrot (Neophema chrysogastor): A key threatening process or an example of a host-adapted virus? PLoS ONE 2019, 14, e0208674. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.; Tse, H.; Chan, W.M.; Choi, G.K.; Zhang, A.J.; Sridhar, S.; Wong, S.C.; Chan, J.F.; Chan, A.S.; Woo, P.C.; et al. A novel psittacine adenovirus identified during an outbreak of avian chlamydiosis and human psittacosis: Zoonosis associated with virus-bacterium coinfection in birds. PLoS Negl. Trop. Dis. 2014, 8, e3318. [Google Scholar] [CrossRef]
- Ballmann, M.Z.; Harrach, B. Detection and partial genetic characterisation of novel avi- and siadenoviruses in racing and fancy pigeons (Columba livia domestica). Acta Vet. Hung. 2016, 64, 514–528. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, E.R.; Benko, M. Complete sequence of raptor adenovirus 1 confirms the characteristic genome organization of siadenoviruses. Infect. Genet. Evol. 2011, 11, 1058–1065. [Google Scholar] [CrossRef]
- Singh, A.K.; Berbis, M.A.; Ballmann, M.Z.; Kilcoyne, M.; Menendez, M.; Nguyen, T.H.; Joshi, L.; Canada, F.J.; Jimenez-Barbero, J.; Benko, M.; et al. Structure and Sialyllactose Binding of the Carboxy-Terminal Head Domain of the Fibre from a Siadenovirus, Turkey Adenovirus 3. PLoS ONE 2015, 10, e0139339. [Google Scholar] [CrossRef]
- Joseph, H.M.; Ballmann, M.Z.; Garner, M.M.; Hanley, C.S.; Berlinski, R.; Erdelyi, K.; Childress, A.L.; Fish, S.S.; Harrach, B.; Wellehan, J.F., Jr. A novel siadenovirus detected in the kidneys and liver of Gouldian finches (Erythura gouldiae). Vet. Microbiol. 2014, 172, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Das, S.; Lavers, J.L.; Hutton, I.; Helbig, K.; Imbery, J.; Upton, C.; Raidal, S.R. Genomic characterization of two novel pathogenic avipoxviruses isolated from pacific shearwaters (Ardenna spp.). BMC Genom. 2017, 18, 298. [Google Scholar] [CrossRef] [Green Version]
- Sarker, S.; Roberts, H.K.; Tidd, N.; Ault, S.; Ladmore, G.; Peters, A.; Forwood, J.K.; Helbig, K.; Raidal, S.R. Molecular and microscopic characterization of a novel Eastern grey kangaroopox virus genome directly from a clinical sample. Sci. Rep. 2017, 7, 16472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athukorala, A.; Forwood, J.K.; Phalen, D.N.; Sarker, S. Molecular Characterisation of a Novel and Highly Divergent Passerine Adenovirus 1. Viruses 2020, 12, 1036. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Altschu, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2014, 42, D32–D37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Tusnady, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef]
- Hofmann, K.; Stoffel, W. Tmbase-A Database of Membrane Spanning Protein Segments. Biol. Chem. 1993, 374, 166. [Google Scholar]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [PubMed]
- ICTV. Famiy—Adenoviridae. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 24 July 2021).
- Penzes, J.J.; Szirovicza, L.; Harrach, B. The complete genome sequence of bearded dragon adenovirus 1 harbors three genes encoding proteins of the C-type lectin-like domain superfamily. Infect. Genet. Evol. 2020, 83, 104321. [Google Scholar] [CrossRef]
- Awadalla, P. The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 2003, 4, 50–60. [Google Scholar] [CrossRef]
- Sarker, S.; Patterson, E.I.; Peters, A.; Baker, B.G.; Forwood, J.K.; Ghorashi, S.A.; Holdsworth, M.; Baker, R.; Murray, N.; Raidal, S.R. Mutability dynamics of an emergent single stranded DNA virus in a naïve host. PLoS ONE 2014, 9, e85370. [Google Scholar]
- Sarker, S.; Ghorashi, S.A.; Forwood, J.K.; Bent, J.S.; Peters, A.; Raidal, S.R. Phylogeny of beak and feather disease virus in cockatoos demonstrates host generalism and multiple-variant infections within Psittaciformes. Virology 2014, 460–461, 72–82. [Google Scholar] [CrossRef]
- Das, S.; Fearnside, K.; Sarker, S.; Forwood, J.K.; Raidal, S.R. A novel pathogenic aviadenovirus from red-bellied parrots (Poicephalus rufiventris) unveils deep recombination events among avian host lineages. Virology 2017, 502, 188–197. [Google Scholar] [CrossRef]
- Froissart, R.; Roze, D.; Uzest, M.; Galibert, L.; Blanc, S.; Michalakis, Y. Recombination Every Day: Abundant Recombination in a Virus during a Single Multi-Cellular Host Infection. PLoS Biol. 2005, 3, e89. [Google Scholar] [CrossRef] [Green Version]
- Lefeuvre, P.; Moriones, E. Recombination as a motor of host switches and virus emergence: Geminiviruses as case studies. Curr. Opin. Virol. 2015, 10, 14–19. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Weiller, G.F. Evidence that a plant virus switched hosts to infect a vertebrate and then recombined with a vertebrate-infecting virus. Proc. Natl. Acad. Sci. USA 1999, 96, 8022–8027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.S.; Eden, J.S.; Hall, J.; Shi, M.; Rose, K.; Holmes, E.C. Metatranscriptomic Analysis of Virus Diversity in Urban Wild Birds with Paretic Disease. J. Virol. 2020, 94, e00606-20. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| PsSiAdV-F Synteny | Start (nt) | Stop (nt) | Strand | Size (aa) | Synteny to PsAdV-D/TAdV-A * | Identity (%) |
|---|---|---|---|---|---|---|
| ORF01 sialidase | 108 | 1970 | + | 620 | Sialidase | 40.32 |
| ORF02 hypothetical protein | 1983 | 2312 | + | 109 | hypothetical protein | 40.48 |
| ORF03 IVa2 | 3429 | 2341 | − | 362 | IVa2 * | 67.40 |
| ORF04 DNA polymerase | 6742 | 3419 | − | 1107 | DNA polymerase | 60.95 |
| ORF05 pTP | 8472 | 6739 | − | 577 | pTP | 59.79 |
| ORF06 52K | 8551 | 9423 | + | 290 | 52K | 71.76 |
| ORF07 pIIIa | 9413 | 10,909 | + | 498 | pIIIa | 60.89 |
| ORF08 penton | 10,931 | 12,271 | + | 446 | Penton | 68.78 |
| ORF09 pVII | 12,272 | 12,661 | + | 129 | pVII | 66.67 |
| ORF10 pX | 12,663 | 12,839 | + | 58 | pX | 67.86 |
| ORF11 pVI | 12,857 | 13,507 | + | 216 | pVI | 52.44 |
| ORF12 hexon | 13,517 | 16,285 | + | 922 | Hexon | 73.65 |
| ORF13 protease | 16,282 | 16,902 | + | 206 | Protease | 60.89 |
| ORF14 early E2 | 17,984 | 16,932 | − | 350 | early E2 | 60.86 |
| ORF15 100K | 18,189 | 20,183 | + | 664 | 100K | 56.04 |
| ORF16 22K | 20,074 | 20,367 | + | 97 | 22K | 71.01 |
| ORF17 33K | 20,386 | 20,667 | + | 93 | 33K * | 70.00 |
| ORF18 pVIII | 20,728 | 21,189 | + | 153 | pVIII | 43.30 |
| ORF19 E3 | 21,137 | 21,706 | + | 189 | E3 | 31.82 |
| ORF20 U exon | 21,971 | 21,753 | − | 72 | U exon | 42.50 |
| ORF21 fiber protein | 21,970 | 23,001 | + | 343 | fiber protein | 35.29 |
| ORF22 hypothetical protein | 23,118 | 23,795 | + | 225 | hypothetical protein | 40.10 |
| ORF23 hypothetical protein | 24,330 | 23,806 | − | 174 | hypothetical protein | 43.04 |
| ORF24 hypothetical protein | 24,578 | 24,330 | − | 82 | hypothetical protein | 42.86 |
| ORF25 hypothetical protein | 25,373 | 24,846 | − | 175 | hypothetical protein | 27.38 |
| Siadenovirus | Genome Identity (%) | G + C Content | % Pairwise AA Identities with PsSiAdV-F | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sialidase | IVa2 | DNA pol | pTP | Penton | Hexon | Protease | DBP | Fiber | |||
| Psittacine siadenovirus F [MW365934] | 36.9 | ||||||||||
| Penguin siadenovirus A [KP144329] | 53.67 | 35.6 | NA | 66.67 | 61.26 | 60.38 | 70.02 | 70.24 | 61.08 | 60.97 | 23.82 |
| Turkey siadenovirus A [AC_000016] | 54.56 | 34.9 | 34.40 | 66.67 | 60.34 | 57.17 | 65.26 | 69.99 | 61.58 | 59.94 | 19.87 |
| Skua siadenovirus A [HM585353] | 57.06 | 34.2 | 44.44 | 66.58 | 61.84 | 57.93 | 69.93 | 72.22 | 58.62 | 61.43 | 19.05 |
| Raptor siadenovirus A [EU715130] | 55.61 | 38.5 | 39.51 | 65.11 | 60.40 | 59.93 | 70.98 | 73.79 | 62.56 | 58.18 | 18.38 |
| Psittacine siadenovirus E [MK227353] | 54.00 | 37.4 | 38.16 | 63.29 | 58.99 | 60.69 | 67.86 | 73.14 | 62.75 | 58.86 | 14.06 |
| Psittacine siadenovirus D [MN687905] | 31.11 | 36.9 | 39.50 | 64.93 | 59.98 | 60.41 | 68.30 | 73.51 | 60.29 | 60.86 | 14.72 |
| Psittacine siadenovirus D [MK695679] | 54.45 | 36.9 | 38.10 | 64.93 | 60.25 | 60.59 | 68.75 | 72.76 | 61.77 | 60.57 | 14.78 |
| Frog siadenovirus A [NC_002501] | 49.47 | 37.9 | 36.96 | 51.91 | 51.57 | 46.65 | 60.54 | 67.02 | 54.68 | 50.28 | 20.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athukorala, A.; Phalen, D.N.; Das, A.; Helbig, K.J.; Forwood, J.K.; Sarker, S. Genomic Characterisation of a Highly Divergent Siadenovirus (Psittacine Siadenovirus F) from the Critically Endangered Orange-Bellied Parrot (Neophema chrysogaster). Viruses 2021, 13, 1714. https://doi.org/10.3390/v13091714
Athukorala A, Phalen DN, Das A, Helbig KJ, Forwood JK, Sarker S. Genomic Characterisation of a Highly Divergent Siadenovirus (Psittacine Siadenovirus F) from the Critically Endangered Orange-Bellied Parrot (Neophema chrysogaster). Viruses. 2021; 13(9):1714. https://doi.org/10.3390/v13091714
Chicago/Turabian StyleAthukorala, Ajani, David N. Phalen, Ashutosh Das, Karla J. Helbig, Jade K. Forwood, and Subir Sarker. 2021. "Genomic Characterisation of a Highly Divergent Siadenovirus (Psittacine Siadenovirus F) from the Critically Endangered Orange-Bellied Parrot (Neophema chrysogaster)" Viruses 13, no. 9: 1714. https://doi.org/10.3390/v13091714