
Visualization of Marek’s Disease Virus Genomes in Living Cells during Lytic Replication and Latency

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Generation of Recombinant Viruses
2.3. Southern Blotting
2.4. Illumina MiSeq Sequencing
2.5. Plaque Size Assay and Growth Kinetics
2.6. Assessment of TetO Stability by Nanopore Sequencing
2.7. Generation of T Cell Line Stably Expressing TetR-mCherry
2.8. Wide-Field Microscopy
2.9. Confocal Microscopy and Live-Cell Imaging
2.10. Lymphocyte Infection
2.11. Fluorescence In Situ Hybridization (FISH)
2.12. Statistical Analysis
3. Results
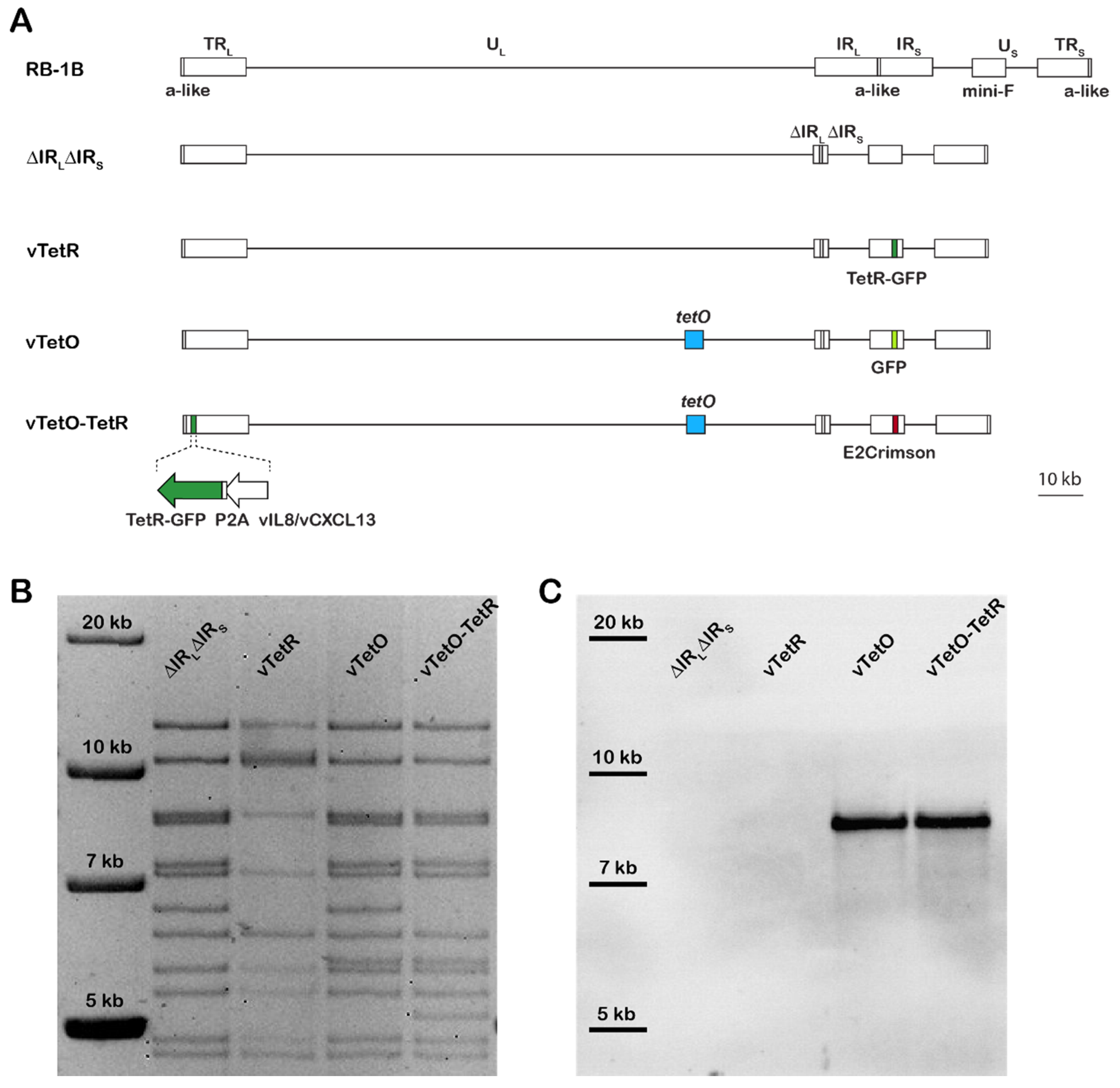
3.1. Generation of Recombinant Viruses
3.2. Characterization of Replication Properties
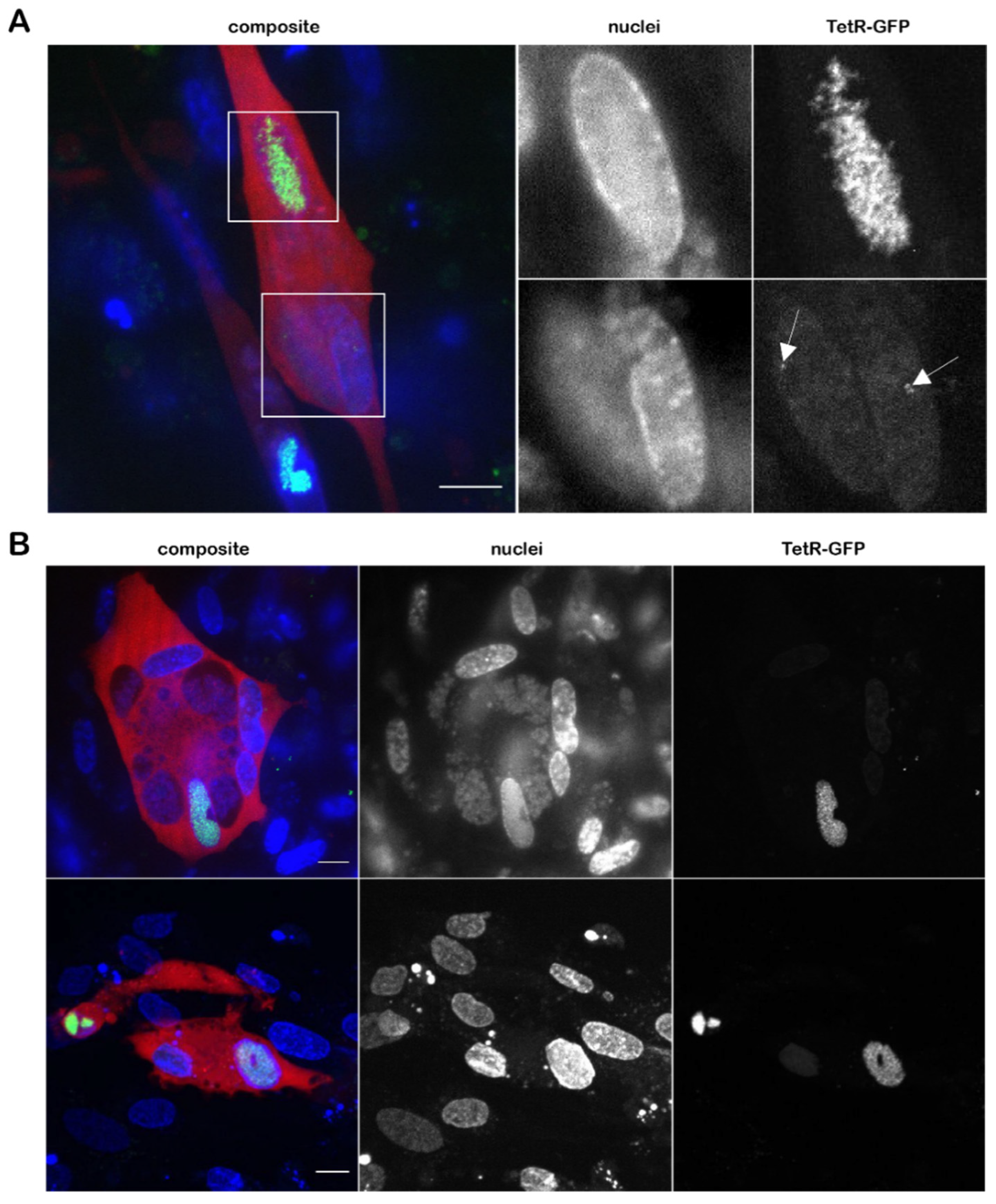
3.3. Visualization of the Virus Genome during Lytic Replication and Specificity of TetR Binding
3.4. Genesis and Mobility of Replication Compartments
3.5. Detection of Both Infected and Uninfected Nuclei inside A Syncytium
3.6. Visualization of Viral Genomes during Lytic Replication and Latency in T Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schat, K.A.; Nair, V. Neoplastic diseases. In Diseases of Poultry, 13th ed.; Swayne, D.E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 513–673. [Google Scholar]
- Barrow, A.; Burgess, S.C.; Baigent, S.J.; Howes, K.; Nair, V. Infection of macrophages by a lymphotropic herpesvirus: A new tropism for Marek’s disease virus. J. Gen. Virol. 2003, 84, 2635–2645. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Vervelde, L.; Dalziel, R.G.; Wasson, P.S.; Nair, V.; Dutia, B.M.; Kaiser, P. Marek’s disease virus infection of phagocytes: A de novo in vitro infection model. J. Gen. Virol. 2017, 98, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Bertzbach, L.D.; Laparidou, M.; Härtle, S.; Etches, R.J.; Kaspers, B.; Schusser, B.; Kaufer, B.B. Unraveling the role of B cells in the pathogenesis of an oncogenic avian herpesvirus. Proc. Natl. Acad. Sci. USA 2018, 115, 11603–11607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertzbach, L.D.; Van Haarlem, D.A.; Härtle, S.; Kaufer, B.B.; Jansen, C.A.; Haarlem, V. Marek’s Disease Virus Infection of Natural Killer Cells. Microorganisms 2019, 7, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermuly, J.; Greco, A.; Härtle, S.; Osterrieder, N.; Kaufer, B.B.; Kaspers, B. In vitro model for lytic replication, latency, and transformation of an oncogenic alphaherpesvirus. Proc. Natl. Acad. Sci. USA 2015, 112, 7279–7284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosinski, K.W.; Tischer, B.K.; Trapp, S.; Osterrieder, N. Marek’s disease virus: Lytic replication, oncogenesis and control. Expert Rev. Vaccines 2006, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Schat, K.A.; Chen, C.L.; Calnek, B.W.; Char, D. Transformation of T-lymphocyte subsets by Marek’s disease herpesvirus. J. Virol. 1991, 65, 1408–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufer, B.B.; Jarosinski, K.W.; Osterrieder, N. Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J. Exp. Med. 2011, 208, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Fester, N.; Engel, A.T.; Kaufer, B.B. Role of the Short Telomeric Repeat Region in Marek’s Disease Virus Replication, Genomic Integration, and Lymphomagenesis. J. Virol. 2014, 88, 14138–14147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parcells, M.S.; Arumugaswami, V.; Prigge, J.T.; Pandya, K.; Dienglewicz, R.L. Marek’s disease virus reactivation from latency: Changes in gene expression at the origin of replication. Poult. Sci. 2003, 82, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Parcells, M.S.; Lin, S.-F.; Dienglewicz, R.L.; Majerciak, V.; Robinson, D.R.; Chen, H.-C.; Wu, Z.; Dubyak, G.R.; Brunovskis, P.; Hunt, H.D.; et al. Marek’s Disease Virus (MDV) Encodes an Interleukin-8 Homolog (vIL-8): Characterization of the vIL-8 Protein and a vIL-8 Deletion Mutant MDV. J. Virol. 2001, 75, 5159–5173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufer, B.B.; Trapp, S.; Jarosinski, K.W.; Osterrieder, N. Herpesvirus Telomerase RNA(vTR)-Dependent Lymphoma Formation Does Not Require Interaction of vTR with Telomerase Reverse Transcriptase (TERT). PLoS Pathog. 2010, 6, e1001073. [Google Scholar] [CrossRef] [PubMed]
- Kheimar, A.; Previdelli, R.L.; Wight, D.J.; Kaufer, B.B. Telomeres and Telomerase: Role in Marek’s Disease Virus Pathogenesis, Integration and Tumorigenesis. Viruses 2017, 9, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mwangi, W.N.; Smith, L.P.; Baigent, S.J.; Beal, R.K.; Nair, V.; Smith, A.L. Clonal Structure of Rapid-Onset MDV-Driven CD4+ Lymphomas and Responding CD8+ T Cells. PLoS Pathog. 2011, 7, e1001337. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, K.; Cui, H.; Gao, L.; Liu, C.; Zhang, Y.; Gao, Y.; Wang, X. Comparison of different sites in recombinant Marek’s disease virus for the expression of green fluorescent protein. Virus Res. 2017, 235, 82–85. [Google Scholar] [CrossRef]
- Denesvre, C.; Rémy, S.; Trapp-Fragnet, L.; Smith, L.P.; Georgeault, S.; Vautherot, J.-F.; Nair, V. Marek’s disease virus undergoes complete morphogenesis after reactivation in a T-lymphoblastoid cell line transformed by recombinant fluorescent marker virus. J. Gen. Virol. 2016, 97, 480–486. [Google Scholar] [CrossRef]
- Jarosinski, K.W.; Donovan, K.M.; Du, G. Expression of fluorescent proteins within the repeat long region of the Marek’s disease virus genome allows direct identification of infected cells while retaining full pathogenicity. Virus Res. 2015, 201, 50–60. [Google Scholar] [CrossRef]
- Rémy, S.; Blondeau, C.; Le Vern, Y.; Lemesle, M.; Vautherot, J.-F.; Denesvre, C. Fluorescent tagging of VP22 in N-terminus reveals that VP22 favors Marek’s disease virus (MDV) virulence in chickens and allows morphogenesis study in MD tumor cells. Vet. Res. 2013, 44, 125. [Google Scholar] [CrossRef] [Green Version]
- Mao, W.; Kim, T.; Cheng, H.H. Visualization of Marek’s disease virus in vitro using enhanced green fluorescent protein fused with US10. Virus Genes 2013, 47, 181–183. [Google Scholar] [CrossRef]
- Jarosinski, K.W.; Arndt, S.; Kaufer, B.B.; Osterrieder, N. Fluorescently Tagged pUL47 of Marek’s Disease Virus Reveals Differential Tissue Expression of the Tegument Protein In Vivo. J. Virol. 2011, 86, 2428–2436. [Google Scholar] [CrossRef] [Green Version]
- Prigge, J.T.; Majerciak, V.; Hunt, H.D.; Dienglewicz, R.L.; Parcells, M.S. Construction and Characterization of Marek’s Disease Viruses Having Green Fluorescent Protein Expression Tied Directly or Indirectly to Phosphoprotein 38 Expression. Avian Dis. 2004, 48, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, J.; Lorenz, A.; Loidl, J. Chromosome associations in budding yeast caused by integrated tandemly repeated transgenes. J. Cell Sci. 2002, 115, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Chang, F.S.; Kleckner, N. Protein-Mediated Chromosome Pairing of Repetitive Arrays. J. Mol. Biol. 2014, 426, 550–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, C.; Ciosk, R.; Nasmyth, K. Cohesins: Chromosomal Proteins that Prevent Premature Separation of Sister Chromatids. Cell 1997, 91, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [PubMed]
- Schat, K.A.; Purchase, H.G.; Amer Assn of Avian Pathologist. Cell-culture methods. In A Laboratory Manual for the Isolation and Identification of Avian Pathogens, 4th ed.; Swayne, D.E., Ed.; American Association of Avian Pathologists: Kennett Square, PA, USA, 1998. [Google Scholar]
- Vautherot, J.-F.; Jean, C.; Fragnet-Trapp, L.; Rémy, S.; Chabanne-Vautherot, D.; Montillet, G.; Fuet, A.; Denesvre, C.; Pain, B. ESCDL-1, a new cell line derived from chicken embryonic stem cells, supports efficient replication of Mardiviruses. PLoS ONE 2017, 12, e0175259. [Google Scholar] [CrossRef]
- Petherbridge, L.; Brown, A.C.; Baigent, S.J.; Howes, K.; Sacco, M.A.; Osterrieder, N.; Nair, V.K. Oncogenicity of Virulent Marek’s Disease Virus Cloned as Bacterial Artificial Chromosomes. J. Virol. 2004, 78, 13376–13380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischer, B.K.; Kaufer, B.B. Viral Bacterial Artificial Chromosomes: Generation, Mutagenesis, and Removal of Mini-F Sequences. J. Biomed. Biotechnol. 2012, 2012, 472537. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Schumacher, D.; Tischer, B.K.; Fuchs, W.; Osterrieder, N. Reconstitution of Marek’s Disease Virus Serotype 1 (MDV-1) from DNA Cloned as a Bacterial Artificial Chromosome and Characterization of a Glycoprotein B-Negative MDV-1 Mutant. J. Virol. 2000, 74, 11088–11098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosinski, K.W.; Margulis, N.G.; Kamil, J.P.; Spatz, S.J.; Nair, V.K.; Osterrieder, N. Horizontal transmission of Marek’s disease virus requires US2, the UL13 protein kinase, and gC. J. Virol. 2007, 81, 10575–10587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trimpert, J.; Groenke, N.; Kunec, D.; Eschke, K.; He, S.; McMahon, D.; Osterrieder, N. A proofreading-impaired herpesvirus generates populations with quasispecies-like structure. Nat. Microbiol. 2019, 4, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Hirt, B. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 1967, 26, 365–369. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Roukos, V.; Voss, T.C.; Schmidt, C.K.; Lee, S.; Wangsa, D.; Misteli, T. Spatial Dynamics of Chromosome Translocations in Living Cells. Science 2013, 341, 660–664. [Google Scholar] [CrossRef]
- Kaufer, B.B. Detection of Integrated Herpesvirus Genomes by Fluorescence In Situ Hybridization (FISH). Program. Necrosis 2013, 1064, 141–152. [Google Scholar] [CrossRef]
- You, Y.; Vychodil, T.; Aimola, G.; Previdelli, R.L.; Göbel, T.W.; Bertzbach, L.D.; Kaufer, B.B. A Cell Culture System to Investigate Marek’s Disease Virus Integration into Host Chromosomes. Microorganisms 2021, 9, 2489. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.M. Visualizing Chromatin Dynamics in Interphase Nuclei. Science 2002, 296, 1412–1416. [Google Scholar] [CrossRef] [PubMed]
- Orth, P.; Cordes, F.; Schnappinger, D.; Hillen, W.; Saenger, W.; Hinrichs, W. Conformational changes of the Tet repressor induced by tetracycline trapping. J. Mol. Biol. 1998, 279, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Seyffert, M.; Georgi, F.; Tobler, K.; Bourqui, L.; Anfossi, M.; Michaelsen, K.; Vogt, B.; Greber, U.; Fraefel, C. The HSV-1 Transcription Factor ICP4 Confers Liquid-Like Properties to Viral Replication Compartments. Int. J. Mol. Sci. 2021, 22, 4447. [Google Scholar] [CrossRef]
- Gupta, M.; Deka, D. Ramneek Sequence analysis of Meq oncogene among Indian isolates of Marek’s disease herpesvirus. Meta Gene 2016, 9, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Boodhoo, N.; Gurung, A.; Sharif, S.; Behboudi, S. Marek’s disease in chickens: A review with focus on immunology. Vet. Res. 2016, 47, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Jarosinski, K.W.; Schat, K.A. Multiple alternative splicing to exons II and III of viral interleukin-8 (vIL-8) in the Marek’s disease virus genome: The importance of vIL-8 exon I. Virus Genes 2007, 34, 9–22. [Google Scholar] [CrossRef]
- Sekine, E.; Schmidt, N.; Gaboriau, D.; O’Hare, P. Spatiotemporal dynamics of HSV genome nuclear entry and compaction state transitions using bioorthogonal chemistry and super-resolution microscopy. PLoS Pathog. 2017, 13, e1006721. [Google Scholar] [CrossRef] [Green Version]
- Dembowski, J.A.; DeLuca, N.A. Temporal Viral Genome-Protein Interactions Define Distinct Stages of Productive Herpesviral Infection. mBio 2018, 9, e01182-18. [Google Scholar] [CrossRef] [Green Version]
- Tomer, E.; Cohen, E.M.; Drayman, N.; Afriat, A.; Weitzman, M.D.; Zaritsky, A.; Kobiler, O. Coalescing replication compartments provide the opportunity for recombination between coinfecting herpesviruses. FASEB J. 2019, 33, 9388–9403. [Google Scholar] [CrossRef]
- Jain, N.; Iyer, K.V.; Kumar, A.; Shivashankar, G.V. Cell geometric constraints induce modular gene-expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proc. Natl. Acad. Sci. USA 2013, 110, 11349–11354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsos, M.; Torres-Padilla, M.-E. Building up the nucleus: Nuclear organization in the establishment of totipotency and pluripotency during mammalian development. Genes Dev. 2016, 30, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Skinner, B.M.; Johnson, E.E.P. Nuclear morphologies: Their diversity and functional relevance. Chromosoma 2017, 126, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McSwiggen, D.T.; Hansen, A.S.; Teves, S.S.; Marie-Nelly, H.; Hao, Y.; Heckert, A.B.; Umemoto, K.K.; Dugast-Darzacq, C.; Tjian, R.; Darzacq, X. Evidence for DNA-mediated nuclear compartmentalization distinct from phase separation. eLife 2019, 8, e47098. [Google Scholar] [CrossRef] [PubMed]
- Sourvinos, G. Visualization of parental HSV-1 genomes and replication compartments in association with ND10 in live infected cells. EMBO J. 2002, 21, 4989–4997. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct/Steps | Direction | Sequence (5′–3′) |
|---|---|---|
| pCDNA3.1 TetR-GFP | for | TAGATGAGCTCGGATCCATGCCAAAGAAGAAGCGTAAG |
| rev | GATGGATATCTGCAGAATTCTCATCCCATGCCATTGGT | |
| TetR-GFP-kana transfer | for | TACAAGACACGTGCTGAAGTCAAGTTTGAAGGTAGGGATAACAGGGTAATCGATTT |
| rev | ACTTCAGCACGTGTCTTGTAGTTCCCGTCATCGCCAGTGTTACAACCAATTAACC | |
| TetR-GFP in mini-F (vTetR) | ep for | TTAAGGTGACACGCGCGGCCTCGAACACAGCTGCAGGCCGATGTACGGGCCAGATATACG |
| ep rev | CGTCGACCCGGGTACCTCTAGATCCGCTAGCGCTTTATGTCTTCCCAATCCTCCCC | |
| P2A-TetR-GFP into vIL-8/vCXCL13 | ep for | ATTGAGCCCACACCTCCTACTATTGGTTCCCATATCTGTCTTGGTTCCGGAGCCACGAACTTCTCTCTGTTAAAGCAAGCAGGAGACGTGGAAGAAAACCCCGGTCCTATGCCAAAGAAGAAGCGTAAG |
| ep rev | AAAGTGCCTTCTTTTAATTACAGGAGGTAGCAATTAATCATCCCATGCCATTGGTAATCC | |
| TetO-kana transfer | for | GACAGTAGATCTACCTGTATACTACCCACCATTGTAGGGATAACAGGGTAATCGATTT |
| rev | ACAGGTAGATCTACTGTCCCGTAGTCTAAATATGCCAGTGTTACAACCAATTAACC | |
| eGFP in mini-F | ep for | GGTGACACGCGCGGCCTCGAACACAGCTGCAGGCCATGGTGAGCAAGGGCGAGG |
| ep rev | CGTCGACCCGGGTACCTCTAGATCCGCTAGCGCTTTACTTGTACAGCTCGTCCATGCC | |
| E2-Crimson into mini-F | ep for | TGCCCTTGCTAGGGTTCTTCACACGAGCCTCGCCTTATTAAATGGGCTCCGGTGCCCGTC |
| ep rev | CCCGAGGCCTCGTGGGGCACCTATTTGCGCGGAGGAAGGCCCATAGAGCCCGGGCCATC | |
| TetO DIG-probe | DIG-TCCCTATCAGTCATAGAGAAAAGTGAAAGTCGAGTTTACCAC | |
| iNOS (qPCR) | for | GAGTGGTTTAAGGAGTTGGATCTGA |
| rev | TTCCAGACCTCCCACCTCAA | |
| probe | FAM-CTCTGCCTGCTGTTGCCAACATGC-TAMRA | |
| UL30 (qPCR) | for | AAGCGGAATCGGTTTACAAG |
| rev | GGAGTTGCTGTTAGAATACGGA | |
| probe | FAM-TCGACGAGTTTCTTCCTCCTCGTTG-TAMRA | |
| Mycoplasma test | for | GGGAGCAAACAGGATTAGATACCCT |
| rev | TGCACCATCTGTCACTCTGTTAACCTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vychodil, T.; Wight, D.J.; Nascimento, M.; Jolmes, F.; Korte, T.; Herrmann, A.; Kaufer, B.B. Visualization of Marek’s Disease Virus Genomes in Living Cells during Lytic Replication and Latency. Viruses 2022, 14, 287. https://doi.org/10.3390/v14020287
Vychodil T, Wight DJ, Nascimento M, Jolmes F, Korte T, Herrmann A, Kaufer BB. Visualization of Marek’s Disease Virus Genomes in Living Cells during Lytic Replication and Latency. Viruses. 2022; 14(2):287. https://doi.org/10.3390/v14020287
Chicago/Turabian StyleVychodil, Tereza, Darren J. Wight, Mariana Nascimento, Fabian Jolmes, Thomas Korte, Andreas Herrmann, and Benedikt B. Kaufer. 2022. "Visualization of Marek’s Disease Virus Genomes in Living Cells during Lytic Replication and Latency" Viruses 14, no. 2: 287. https://doi.org/10.3390/v14020287