
The In Vitro Replication, Spread, and Oncolytic Potential of Finnish Circulating Strains of Herpes Simplex Virus Type 1

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Viruses
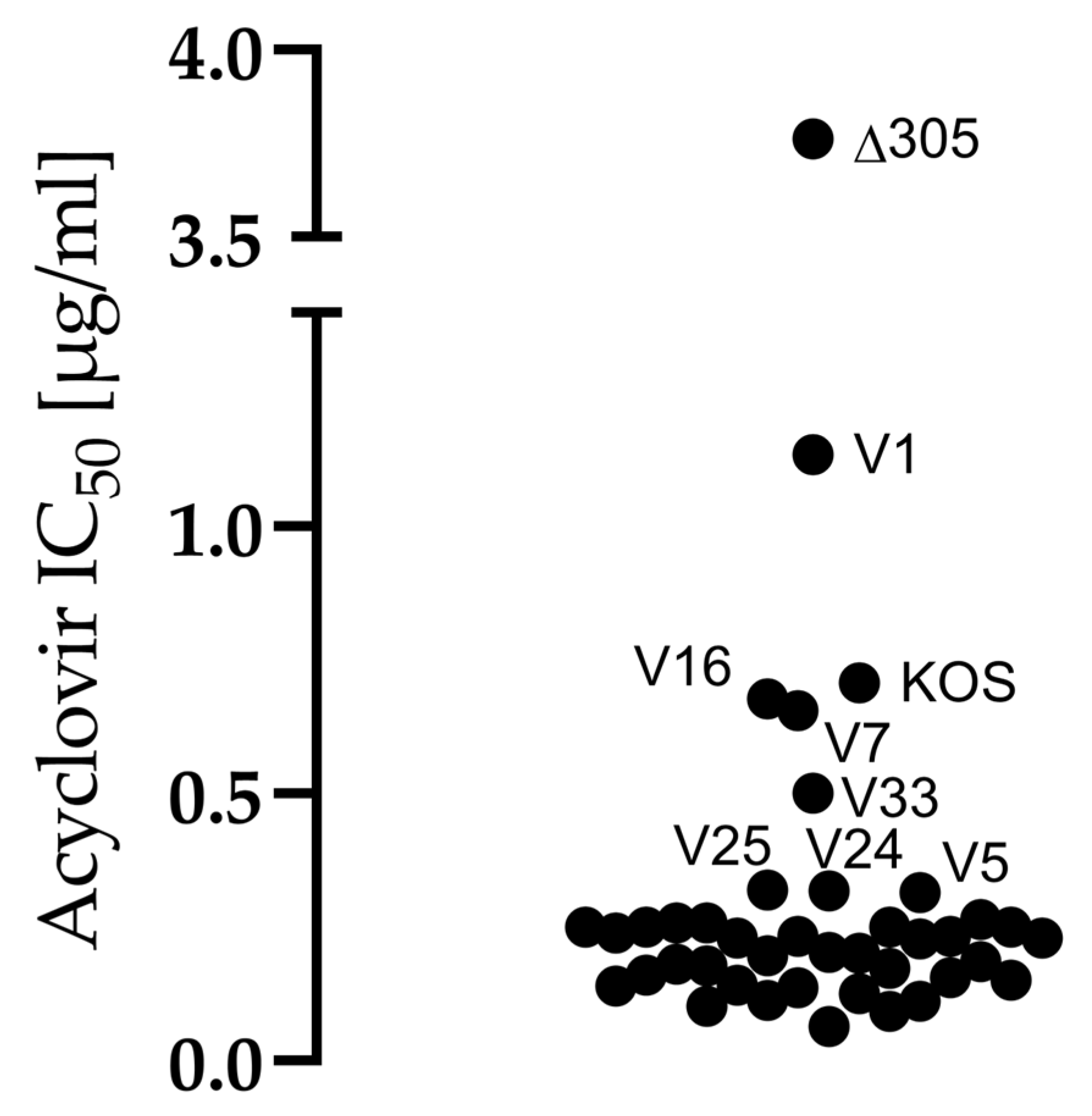
2.3. Acyclovir Sensitivity Assay
2.4. Virus Yield
2.5. Assay Comparing Cell-Associated and Extracellular Virus
2.6. Sequencing
2.7. Cytolytic Assay for Determination of Oncolytic Potential
2.8. Statistical Analysis
3. Results
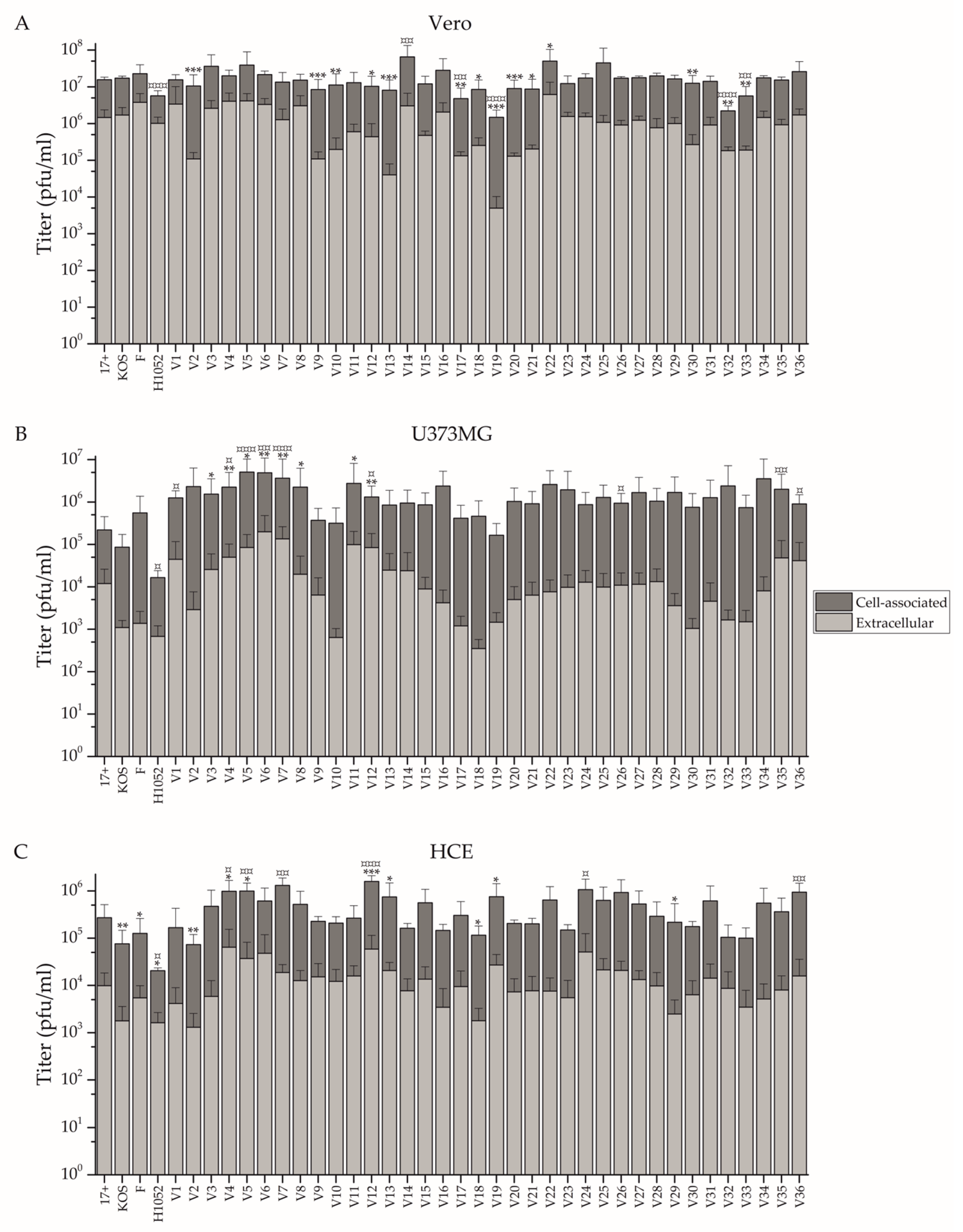
3.1. Comparison of Growth Properties of the Circulating Strains In Vitro
3.2. US7 and UL27 Genotypes of the Clinical Isolates
3.3. Oncolytic Potential
4. Discussion
4.1. Replication and Spread of the Clinical Isolates Is Dependent on the Cell Type
4.2. All Clinical Isolates Were Sensitive to Acyclovir
4.3. The Phenotype Did Not Predict the US7 nor UL27 Genotype
4.4. The Clinical Isolates Could Be Advantageous as oHSV Backbones
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Szpara, M.L.; Parsons, L.; Enquist, L.W. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 2010, 84, 5303–5313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and diversity in human herpes simplex virus genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, C.D.; Paavilainen, H.; Renner, D.W.; Palomaki, J.; Lehtinen, J.; Vuorinen, T.; Norberg, P.; Hukkanen, V.; Szpara, M.L. Comparison of Herpes Simplex Virus 1 Strains Circulating in Finland Demonstrates the Uncoupling of Whole-Genome Relatedness and Phenotypic Outcomes of Viral Infection. J. Virol. 2019, 93, e01824-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norberg, P.; Bergstrom, T.; Rekabdar, E.; Lindh, M.; Liljeqvist, J.A. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J. Virol. 2004, 78, 10755–10764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer 2019, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalke, K.; Lehtinen, J.; Gnjatovic, J.; Lund, L.M.; Nyman, M.C.; Paavilainen, H.; Orpana, J.; Lasanen, T.; Frejborg, F.; Levanova, A.A.; et al. Herpes Simplex Virus Type 1 Clinical Isolates Respond to UL29-Targeted siRNA Swarm Treatment Independent of Their Acyclovir Sensitivity. Viruses 2020, 12, 1434. [Google Scholar] [CrossRef] [PubMed]
- Shipley, M.M.; Renner, D.W.; Ott, M.; Bloom, D.C.; Koelle, D.M.; Johnston, C.; Szpara, M.L. Genome-Wide Surveillance of Genital Herpes Simplex Virus Type 1 From Multiple Anatomic Sites Over Time. J. Infect. Dis. 2018, 218, 595–605. [Google Scholar] [CrossRef]
- Chou, J.; Chen, J.J.; Gross, M.; Roizman, B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 1995, 92, 10516–10520. [Google Scholar]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar]
- Hukkanen, V.; Nygardas, M. The virtues and vices of harnessing HSV vectors for CNS autoimmunity modulation. Immunotherapy 2013, 5, 911–913. [Google Scholar] [CrossRef]
- Roth, J.C.; Cassady, K.A.; Cody, J.J.; Parker, J.N.; Price, K.H.; Coleman, J.M.; Peggins, J.O.; Noker, P.E.; Powers, N.W.; Grimes, S.D.; et al. Evaluation of the safety and biodistribution of M032, an attenuated herpes simplex virus type 1 expressing hIL-12, after intracerebral administration to aotus nonhuman primates. Hum. Gene Ther. Clin. Dev. 2014, 25, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- MacLean, A.R.; ul-Fareed, M.; Robertson, L.; Harland, J.; Brown, S.M. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J. Gen. Virol. 1991, 72 Pt 3, 631–639. [Google Scholar] [CrossRef]
- Eissa, I.R.; Naoe, Y.; Bustos-Villalobos, I.; Ichinose, T.; Tanaka, M.; Zhiwen, W.; Mukoyama, N.; Morimoto, T.; Miyajima, N.; Hitoki, H.; et al. Genomic Signature of the Natural Oncolytic Herpes Simplex Virus HF10 and Its Therapeutic Role in Preclinical and Clinical Trials. Front. Oncol. 2017, 7, 149. [Google Scholar] [CrossRef]
- Cassady, K.A. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 2005, 79, 8707–8715. [Google Scholar] [CrossRef] [Green Version]
- Haines, B.B.; Denslow, A.; Grzesik, P.; Lee, J.S.; Farkaly, T.; Hewett, J.; Wambua, D.; Kong, L.; Behera, P.; Jacques, J.; et al. ONCR-177, an Oncolytic HSV-1 Designed to Potently Activate Systemic Antitumor Immunity. Cancer Immunol. Res. 2021, 9, 291–308. [Google Scholar] [CrossRef]
- Ren, J.; Gwin, W.R.; Zhou, X.; Wang, X.; Huang, H.; Jiang, N.; Zhou, L.; Agarwal, P.; Hobeika, A.; Crosby, E.; et al. Adaptive T cell responses induced by oncolytic Herpes Simplex Virus-granulocyte macrophage-colony-stimulating factor therapy expanded by dendritic cell and cytokine-induced killer cell adoptive therapy. Oncoimmunology 2017, 6, e1264563. [Google Scholar] [CrossRef] [Green Version]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [Green Version]
- Chouljenko, D.V.; Ding, J.; Lee, I.F.; Murad, Y.M.; Bu, X.; Liu, G.; Delwar, Z.; Sun, Y.; Yu, S.; Samudio, I.; et al. Induction of Durable Antitumor Response by a Novel Oncolytic Herpesvirus Expressing Multiple Immunomodulatory Transgenes. Biomedicines 2020, 8, 484. [Google Scholar] [CrossRef]
- Kelly, K.J.; Wong, J.; Fong, Y. Herpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancy. Expert Opin. Investig. Drugs 2008, 17, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.; Sahu, U.; Mullarkey, M.P.; Kaur, B. Replication and Spread of Oncolytic Herpes Simplex Virus in Solid Tumors. Viruses 2022, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Kuny, C.V.; Bowen, C.D.; Renner, D.W.; Johnston, C.M.; Szpara, M.L. In vitro evolution of herpes simplex virus 1 (HSV-1) reveals selection for syncytia and other minor variants in cell culture. Virus Evol. 2020, 6, veaa013. [Google Scholar] [CrossRef]
- Ziegler, T.; Waris, M.; Rautiainen, M.; Arstila, P. Herpes simplex virus detection by macroscopic reading after overnight incubation and immunoperoxidase staining. J. Clin. Microbiol. 1988, 26, 2013–2017. [Google Scholar] [CrossRef] [Green Version]
- Post, L.E.; Mackem, S.; Roizman, B. Regulation of alpha genes of herpes simplex virus: Expression of chimeric genes produced by fusion of thymidine kinase with alpha gene promoters. Cell 1981, 24, 555–565. [Google Scholar] [CrossRef]
- Mattila, R.K.; Harila, K.; Kangas, S.M.; Paavilainen, H.; Heape, A.M.; Mohr, I.J.; Hukkanen, V. An investigation of herpes simplex virus type 1 latency in a novel mouse dorsal root ganglion model suggests a role for ICP34.5 in reactivation. J. Gen. Virol. 2015, 96, 2304–2313. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dolan, A.; Donald, S.; Brauer, D.H. Complete DNA sequence of the short repeat region in the genome of herpes simplex virus type 1. Nucleic Acids Res. 1986, 14, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69 Pt 7, 1531–1574. [Google Scholar] [CrossRef]
- Smith, K.O. Relationship between the Envelope and the Infectivity of Herpes Simplex Virus. Proc. Soc. Exp. Biol. Med. 1964, 115, 814–816. [Google Scholar] [CrossRef]
- Ejercito, P.M.; Kieff, E.D.; Roizman, B. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 1968, 2, 357–364. [Google Scholar] [CrossRef]
- Pan, D.; Kaye, S.B.; Hopkins, M.; Kirwan, R.; Hart, I.J.; Coen, D.M. Common and new acyclovir resistant herpes simplex virus-1 mutants causing bilateral recurrent herpetic keratitis in an immunocompetent patient. J. Infect. Dis. 2014, 209, 345–349. [Google Scholar] [CrossRef]
- Duan, R.; de Vries, R.D.; van Dun, J.M.; van Loenen, F.B.; Osterhaus, A.D.; Remeijer, L.; Verjans, G.M. Acyclovir susceptibility and genetic characteristics of sequential herpes simplex virus type 1 corneal isolates from patients with recurrent herpetic keratitis. J. Infect. Dis. 2009, 200, 1402–1414. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.; van Dun, J.M.; Remeijer, L.; Siemerink, M.; Mulder, P.G.; Norberg, P.; Osterhaus, A.D.; Verjans, G.M. Prevalence of herpes simplex virus type 1 glycoprotein G (gG) and gI genotypes in patients with herpetic keratitis. Br. J. Ophthalmol. 2008, 92, 1195–1200. [Google Scholar] [CrossRef]
- Ishino, R.; Kawase, Y.; Kitawaki, T.; Sugimoto, N.; Oku, M.; Uchida, S.; Imataki, O.; Matsuoka, A.; Taoka, T.; Kawakami, K.; et al. Oncolytic Virus Therapy with HSV-1 for Hematological Malignancies. Mol. Ther. 2021, 29, 762–774. [Google Scholar] [CrossRef]
- Ghose, J.; Dona, A.; Murtadha, M.; Gunes, E.G.; Caserta, E.; Yoo, J.Y.; Russell, L.; Jaime-Ramirez, A.C.; Barwick, B.G.; Gupta, V.A.; et al. Oncolytic herpes simplex virus infects myeloma cells in vitro and in vivo. Mol. Ther. Oncolytics 2021, 20, 519–531. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Parental Strain | Target 1 | Phase | Status | Identifier | Ref. |
|---|---|---|---|---|---|---|
| T-VEC (Imlygic) | JS1 2 | Melanoma | II | Recruiting | NCT04330430 | [5] |
| M032 | F | Glioblastoma | I/II | Not yet recruiting | NCT05084430 | [12] |
| G207 | F | Pediatric brain tumors | II | Not yet recruiting | NCT04482933 | [13] |
| HSV1716 | 17+ | Mesothelioma | I/II | Completed | NCT01721018 | [14] |
| C-REV (HF10) | HF | Melanoma | II | Completed | NCT03153085 | [15] |
| C134 | F | Glioblastoma | I | Recruiting | NCT03657576 | [16] |
| ONCR-177 | KOS | Melanoma | I | Recruiting | NCT04348916 | [17] |
| OrienX010 | CL1 2 | Melanoma | I | Recruiting | NCT04206358 | [18] |
| RP1 | RH018 2 | Melanoma | II | Recruiting | NCT03767348 | [6] |
| RP2 | RH018 2 | Cancer | I | Recruiting | NCT04336241 | [6] |
| RP3 | RH018 2 | Advanced Solid Tumor | I | Recruiting | NCT04735978 | [6] |
| rQNestin | F | Brain cancer | I | Recruiting | NCT03152318 | [19] |
| VG161 | 17+ | Liver cancer | II | Not yet recruiting | NCT05223816 | [20] |
| NV1020 | F | Colorectal cancer | I/II | Completed | NCT00149396 | [21] |
| G47Δ | F | Glioblastoma | II | Completed | UMIN000015995 | [22] |
| Virus | Description | Reference |
|---|---|---|
| V1-V36 | Clinical isolates of herpes simplex virus type 1 (HSV-1) | This paper |
| HSV-1 Δ305 | HSV-1 strain devoid of thymidine kinase | [26] |
| HSV-1 (17+) | HSV-1 reference strain | [28,29] |
| HSV-1 (KOS) | HSV-1 reference strain | [30] |
| HSV-1 (F) | HSV-1 reference strain | [31] |
| H1052 | Derivative of HSV-1 17+ strain with pmCMV-eGFP and phCMV-LUC transgenes and neurovirulence gene (γ134.5) deletions | [27] |
| Vero | U373MG | HCE | Vero | U373MG | HCE | ||
|---|---|---|---|---|---|---|---|
| 17+ | 12.0% | 2.4% | 7.6% | V17 | 6.4% | 0.8% | 3.3% |
| KOS | 11.0% | 0.2% | 3.8% | V18 | 8.4% | 0.1% | 2.1% |
| F | 25.8% | 0.5% | 2.4% | V19 | 0.4% | 2.5% | 5.4% |
| H1052 | 28.9% | 1.7% | 8.6% | V20 | 3.3% | 0.9% | 3.4% |
| V1 | 39.0% | 3.0% | 7.2% | V21 | 4.9% | 0.7% | 3.8% |
| V2 | 2.9% | 0.2% | 2.3% | V22 | 47.4% | 1.0% | 1.6% |
| V3 | 15.2% | 1.5% | 3.8% | V23 | 41.8% | 1.4% | 3.9% |
| V4 | 65.8% | 3.3% | 13.3% | V24 | 12.1% | 2.6% | 4.8% |
| V5 | 18.9% | 1.3% | 4.0% | V25 | 44.8% | 1.1% | 5.5% |
| V6 | 18.3% | 6.0% | 8.3% | V26 | 5.6% | 1.2% | 4.7% |
| V7 | 25.5% | 7.4% | 1.8% | V27 | 7.2% | 1.2% | 4.1% |
| V8 | 90.5% | 1.8% | 3.7% | V28 | 6.0% | 1.4% | 4.7% |
| V9 | 4.7% | 2.4% | 8.0% | V29 | 7.1% | 0.1% | 1.3% |
| V10 | 5.4% | 0.2% | 8.9% | V30 | 3.3% | 0.2% | 3.4% |
| V11 | 10.0% | 6.0% | 7.9% | V31 | 9.2% | 1.7% | 2.7% |
| V12 | 5.5% | 6.7% | 4.6% | V32 | 10.3% | 0.2% | 7.8% |
| V13 | 0.6% | 2.2% | 5.9% | V33 | 6.6% | 0.1% | 4.0% |
| V14 | 8.8% | 1.8% | 5.4% | V34 | 9.6% | 0.6% | 1.7% |
| V15 | 10.4% | 0.7% | 2.9% | V35 | 7.1% | 2.7% | 3.0% |
| V16 | 10.9% | 0.3% | 2.2% | V36 | 10.4% | 3.5% | 1.5% |
| 1 Higher Extracellular Virus Titer Than 17+ | 1 Higher Cell- Associated Titer Than 17+ | 2 Higher Proportional Shedding Than 17+ | 2 Higher Proportional Cell-Associated Virus Than 17+ | 3 More Oncolytic Than 17+ | 3 More Oncolytic Than F | 3 More Oncolytic Than KOS | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| U373MG | HCE | Vero | U373MG | HCE | Vero | U373MG | HCE | Vero | U373MG | HCE | Vero | U373MG | Raji | SW480 | U373MG | Raji | SW480 | U373MG | Raji | SW480 | |
| V1 | X | X | X | X | X | X | |||||||||||||||
| V2 | X | X | X | X | X | X | X | ||||||||||||||
| V3 | X | X | X | X | X | X | |||||||||||||||
| V4 | X | X | X | X | X | X | X | X | X | X | |||||||||||
| V5 | X | X | X | X | X | X | X | X | X | X | |||||||||||
| V6 | X | X | X | X | X | X | X | ||||||||||||||
| V7 | X | X | X | X | X | X | X | X | X | ||||||||||||
| V8 | X | X | X | X | X | X | |||||||||||||||
| V9 | X | X | X | X | |||||||||||||||||
| V10 | X | X | X | ||||||||||||||||||
| V11 | X | X | X | X | |||||||||||||||||
| V12 | X | X | X | X | X | X | X | X | X | X | X | ||||||||||
| V13 | X | X | X | X | X | X | |||||||||||||||
| V14 | X | X | X | X | X | X | |||||||||||||||
| V15 | X | X | X | X | X | ||||||||||||||||
| V16 | X | X | X | X | |||||||||||||||||
| V17 | X | X | X | ||||||||||||||||||
| V18 | X | X | X | ||||||||||||||||||
| V19 | X | X | X | X | X | ||||||||||||||||
| V20 | X | X | X | X | X | X | |||||||||||||||
| V21 | X | X | X | X | |||||||||||||||||
| V22 | X | X | X | X | |||||||||||||||||
| V23 | X | X | X | ||||||||||||||||||
| V24 | X | X | X | X | X | X | |||||||||||||||
| V25 | X | X | X | ||||||||||||||||||
| V26 | X | X | X | X | X | ||||||||||||||||
| V27 | X | X | X | X | X | X | X | X | |||||||||||||
| V28 | X | X | X | X | X | ||||||||||||||||
| V29 | X | X | X | X | X | X | X | ||||||||||||||
| V30 | X | X | X | X | |||||||||||||||||
| V31 | X | X | X | X | X | X | |||||||||||||||
| V32 | X | X | X | X | X | ||||||||||||||||
| V33 | X | X | X | X | X | ||||||||||||||||
| V34 | X | X | X | X | X | ||||||||||||||||
| V35 | X | X | X | X | X | X | X | X | X | ||||||||||||
| V36 | X | X | X | X | X | X | X | X | |||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalke, K.; Orpana, J.; Lasanen, T.; Esparta, O.; Lund, L.M.; Frejborg, F.; Vuorinen, T.; Paavilainen, H.; Hukkanen, V. The In Vitro Replication, Spread, and Oncolytic Potential of Finnish Circulating Strains of Herpes Simplex Virus Type 1. Viruses 2022, 14, 1290. https://doi.org/10.3390/v14061290
Kalke K, Orpana J, Lasanen T, Esparta O, Lund LM, Frejborg F, Vuorinen T, Paavilainen H, Hukkanen V. The In Vitro Replication, Spread, and Oncolytic Potential of Finnish Circulating Strains of Herpes Simplex Virus Type 1. Viruses. 2022; 14(6):1290. https://doi.org/10.3390/v14061290
Chicago/Turabian StyleKalke, Kiira, Julius Orpana, Tuomas Lasanen, Olaya Esparta, Liisa M. Lund, Fanny Frejborg, Tytti Vuorinen, Henrik Paavilainen, and Veijo Hukkanen. 2022. "The In Vitro Replication, Spread, and Oncolytic Potential of Finnish Circulating Strains of Herpes Simplex Virus Type 1" Viruses 14, no. 6: 1290. https://doi.org/10.3390/v14061290