
Chikungunya Virus Asian Lineage Infection in the Amazon Region Is Maintained by Asiatic and Caribbean-Introduced Variants

 ,
,  , , , ,
, , , ,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Processing and Quantitative Real-Time RT-PCR
2.3. Library Preparation and Next-Generation Sequencing
2.4. Phylogenetic and Bayesian Analysis
2.5. Epidemiological Data Compilation
3. Results
3.1. PCR Assay
3.2. Location of Sample Collection
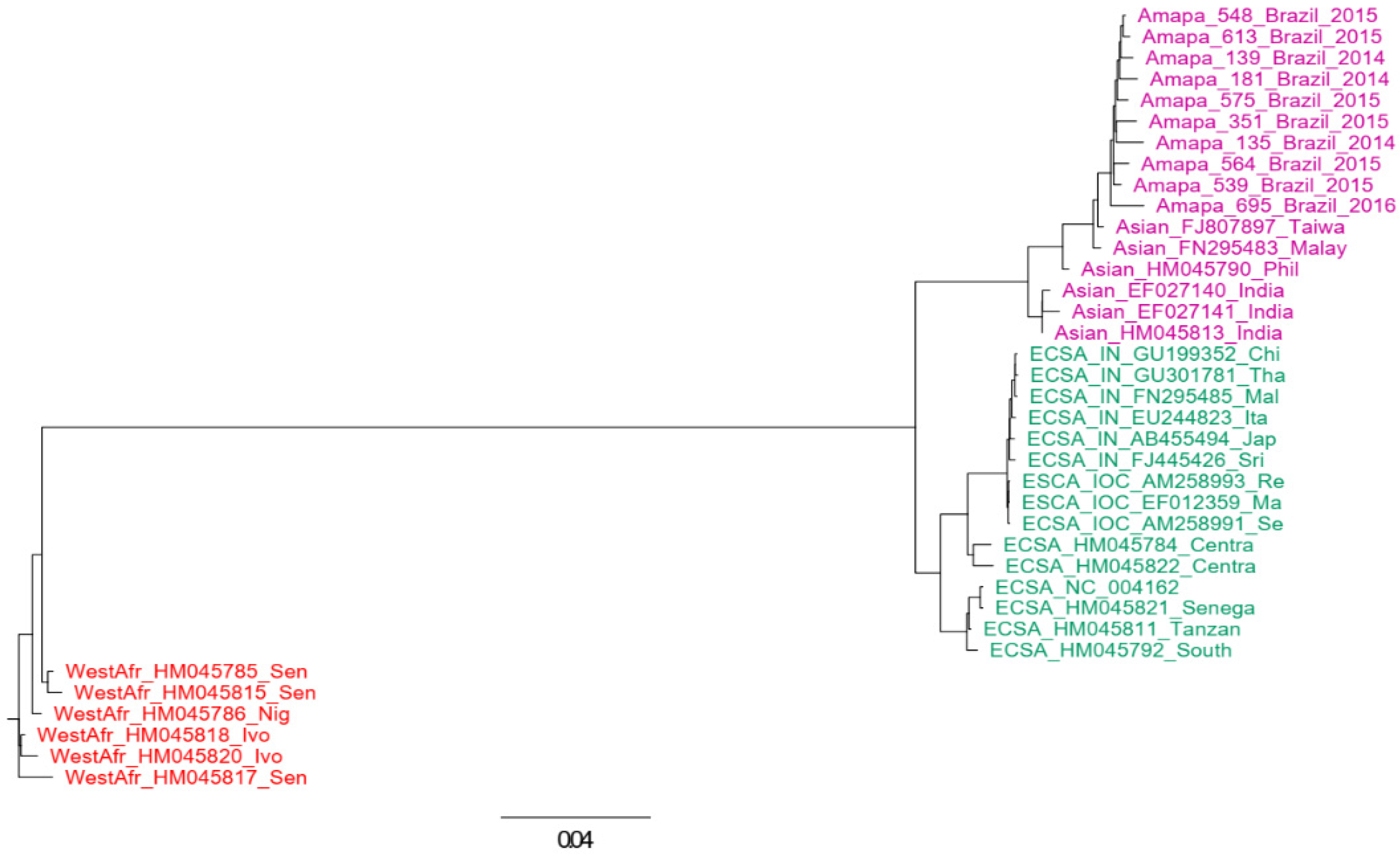
3.3. Next Generation Sequencing and Genotyping Tree
3.4. Phylogenetic Analysis
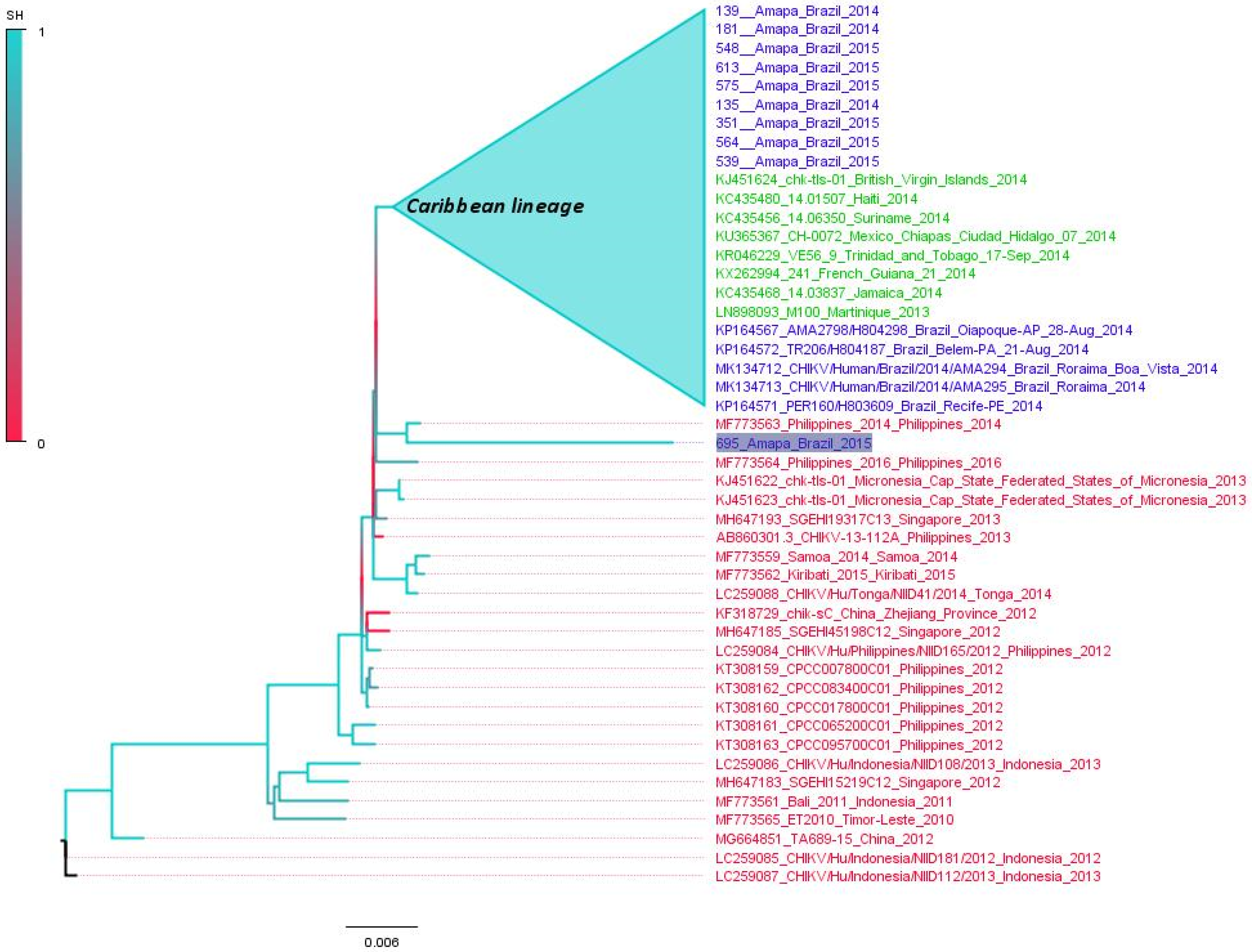
3.5. Polyphyletic versus Monophyletic Pattern of CHIKV from Amapá
3.6. Time-Scaled Tree
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Costa-da-Silva, A.L.; Ioshino, R.S.; Petersen, V.; Lima, A.F.; dos Passos Cunha, M.; Wiley, M.R.; Ladner, J.T.; Prieto, K.; Palacios, G.; Costa, D.D.; et al. First Report of Naturally Infected Aedes aegypti with Chikungunya Virus Genotype ECSA in the Americas. PLoS Negl. Trop. Dis. 2017, 11, e0005630. [Google Scholar] [CrossRef] [PubMed]
- Eskildsen, G.A.; Rovira, J.R.; Smith, O.; Miller, M.J.; Bennett, K.L.; McMillan, W.O.; Loaiza, J. Maternal Invasion History of Aedes aegypti and Aedes albopictus into the Isthmus of Panama: Implications for the Control of Emergent Viral Disease Agents. PLoS ONE 2018, 13, e0194874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega-Rúa, A.; Zouache, K.; Girod, R.; Failloux, A.-B.; Lourenço-de-Oliveira, R. High Level of Vector Competence of Aedes aegypti and Aedes albopictus from Ten American Countries as a Crucial Factor in the Spread of Chikungunya Virus. J. Virol. 2014, 88, 6294–6306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, J.; Fonseca, V.; Bezerra, J.F.; do Monte Alves, M.; Mares-Guia, M.A.; Claro, I.M.; de Jesus, R.; Adelino, T.; Araújo, E.; Cavalcante, K.R.L.J.; et al. Chikungunya Virus ECSA Lineage Reintroduction in the Northeasternmost Region of Brazil. Int. J. Infect. Dis. 2021, 105, 120–123. [Google Scholar] [CrossRef]
- Monteiro, J.D.; Valverde, J.G.; Morais, I.C.; de Medeiros Souza, C.R.; Fagundes Neto, J.C.; de Melo, M.F.; Nascimento, Y.M.; Alves, B.E.B.; de Medeiros, L.G.; Pereira, H.W.B.; et al. Epidemiologic and Clinical Investigations during a Chikungunya Outbreak in Rio Grande Do Norte State, Brazil. PLoS ONE 2020, 15, e0241799. [Google Scholar] [CrossRef]
- Elsinga, J.; Gerstenbluth, I.; van der Ploeg, S.; Halabi, Y.; Lourents, N.T.; Burgerhof, J.G.; van der Veen, H.T.; Bailey, A.; Grobusch, M.P.; Tami, A. Long-Term Chikungunya Sequelae in Curaçao: Burden, Determinants, and a Novel Classification Tool. J. Infect. Dis. 2017, 216, 573–581. [Google Scholar] [CrossRef]
- Yactayo, S.; Staples, J.E.; Millot, V.; Cibrelus, L.; Ramon-Pardo, P. Epidemiology of Chikungunya in the Americas. J. Infect. Dis. 2016, 214, S441–S445. [Google Scholar] [CrossRef] [Green Version]
- Simizu, B.; Yamamoto, K.; Hashimoto, K.; Ogata, T. Structural Proteins of Chikungunya Virus. J. Virol. 1984, 51, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Powers, A.M.; Brault, A.C.; Tesh, R.B.; Weaver, S.C. Re-Emergence of Chikungunya and O’nyong-Nyong Viruses: Evidence for Distinct Geographical Lineages and Distant Evolutionary Relationships. Microbiology 2000, 81, 471–479. [Google Scholar] [CrossRef]
- Robinson, M.C. An Epidemic of Virus Disease in Southern Province, Tanganyika Territory, in 1952–1953. I. Clinical Features. Trans. R. Soc. Trop. Med. Hyg. 1955, 49, 28–32. [Google Scholar] [CrossRef]
- Ross, R.W. The Newala Epidemic: III. The Virus: Isolation, Pathogenic Properties and Relationship to the Epidemic. J. Hyg. 1956, 54, 177–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyango, C.; Breiman, R.F.; Ofula, V.; Bedno, S.; Konongoi, L.S.; Burke, H.; Konde, J.; Sergon, K.; Sang, R.; Dumilla, A.M.; et al. Seroprevalence of Chikungunya Virus (CHIKV) Infection on Lamu Island, Kenya, October 2004. Am. J. Trop. Med. Hyg. 2008, 78, 333–337. [Google Scholar] [CrossRef]
- Gallian, P.; Leparc-Goffart, I.; Richard, P.; Maire, F.; Flusin, O.; Djoudi, R.; Chiaroni, J.; Charrel, R.; Tiberghien, P.; de Lamballerie, X. Epidemiology of Chikungunya Virus Outbreaks in Guadeloupe and Martinique, 2014: An Observational Study in Volunteer Blood Donors. PLoS Negl. Trop. Dis. 2017, 11, e0005254. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.R.T.; Faria, N.R.; de Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.; de Oliveira, L.F.; do Socorro da Silva Azevedo, R.; da Silva, D.E.A.; da Silva, E.V.P.; da Silva, S.P.; et al. Emergence and Potential for Spread of Chikungunya Virus in Brazil. BMC Med. 2015, 13, 102. [Google Scholar] [CrossRef] [Green Version]
- Machado, L.C.; de Morais-Sobral, M.C.; de Lima Campos, T.; Pereira, M.R.; de Fátima Pessoa Militão de Albuquerque, M.; Gilbert, C.; Franca, R.F.O.; Wallau, G.L. Genome Sequencing Reveals Coinfection by Multiple Chikungunya Virus Genotypes in a Recent Outbreak in Brazil. PLoS Negl. Trop. Dis. 2019, 13, e0007332. [Google Scholar] [CrossRef]
- Rodrigues, A.M.; Souza, R.R.M.; dos Santos Fonseca, L.M.; de Araújo Rolo, C.; Carvalho, R.H.; Sardi, S.I.; Campos, G.S. Genomic Surveillance of the Chikungunya Virus (CHIKV) in Northeast Brazil after the First Outbreak in 2014. Rev. Soc. Bras. Med. Trop. 2020, 53, e20190583. [Google Scholar] [CrossRef]
- Conteville, L.C.; Zanella, L.; Marín, M.A.; de Filippis, A.M.B.; Nogueira, R.M.R.; Vicente, A.C.P.; de Mendonça, M.C.L. Phylogenetic Analyses of Chikungunya Virus among Travelers in Rio de Janeiro, Brazil, 2014–2015. Mem. Inst. Oswaldo Cruz 2016, 111, 347–348. [Google Scholar] [CrossRef] [Green Version]
- Naveca, F.G.; Claro, I.; Giovanetti, M.; de Jesus, J.G.; Xavier, J.; de Melo Iani, F.C.; do Nascimento, V.A.; de Souza, V.C.; Silveira, P.P.; Lourenço, J.; et al. Genomic, Epidemiological and Digital Surveillance of Chikungunya Virus in the Brazilian Amazon. PLoS Negl. Trop. Dis. 2019, 13, e0007065. [Google Scholar] [CrossRef] [Green Version]
- Van Bortel, W.; Dorleans, F.; Rosine, J.; Blateau, A.; Rousset, D.; Matheus, S.; Leparc-Goffart, I.; Flusin, O.; Prat, C.M.; Césaire, R.; et al. Chikungunya Outbreak in the Caribbean Region, December 2013 to March 2014, and the Significance for Europe. Eurosurveillance 2014, 19, 20759. [Google Scholar] [CrossRef] [Green Version]
- Charlys da Costa, A.; Thézé, J.; Komninakis, S.C.V.; Sanz-Duro, R.L.; Felinto, M.R.L.; Moura, L.C.C.; Barroso, I.M.d.O.; Santos, L.E.C.; Nunes, M.A.d.L.; Moura, A.A.; et al. Spread of Chikungunya Virus East/Central/South African Genotype in Northeast Brazil. Emerg. Infect. Dis. 2017, 23, 1742–1744. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, V.; Libin, P.J.K.; Theys, K.; Faria, N.R.; Nunes, M.R.T.; Restovic, M.I.; Freire, M.; Giovanetti, M.; Cuypers, L.; Nowé, A.; et al. A Computational Method for the Identification of Dengue, Zika and Chikungunya Virus Species and Genotypes. PLoS Negl. Trop. Dis. 2019, 13, e0007231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, A. AliView: A Fast and Lightweight Alignment Viewer and Editor for Large Datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. JModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, G.d.O.; Gill, D.E.; Ribeiro, E.S.D.; Monteiro, F.J.C.; Morais, V.S.; Marcatti, R.; Rego, M.O.d.S.; Araújo, E.L.L.; Witkin, S.S.; Villanova, F.; et al. Adaptive Evolution of New Variants of Dengue Virus Serotype 1 Genotype V Circulating in the Brazilian Amazon. Viruses 2021, 13, 689. [Google Scholar] [CrossRef]
- Pyke, A.T.; Moore, P.R.; McMahon, J. New Insights into Chikungunya Virus Emergence and Spread from Southeast Asia. Emerg. Microbes Infect. 2018, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Dezécache, C.; Faure, E.; Gond, V.; Salles, J.-M.; Vieilledent, G.; Hérault, B. Gold-Rush in a Forested El Dorado: Deforestation Leakages and the Need for Regional Cooperation. Environ. Res. Lett. 2017, 12, 034013. [Google Scholar] [CrossRef]
- Peiter, P.C. Condiciones de Vida, Situación de la Salud y Disponibilidad de Servicios de Salud en la Frontera de Brasil: Un Enfoque Geográfico. Cad. Saúde Pública 2007, 23, S237–S250. [Google Scholar] [CrossRef] [Green Version]
- Seid, M.; Castañeda, D.; Mize, R.; Zivkovic, M.; Varni, J.W. Crossing the Border for Health Care: Access and Primary Care Characteristics for Young Children of Latino Farm Workers along the US-Mexico Border. Ambul. Pediatr. 2003, 3, 121–130. [Google Scholar] [CrossRef]
- Nunes, M.R.T.; Faria, N.R.; Vasconcelos, H.B.; Medeiros, D.B.d.A.; Silva de Lima, C.P.; Carvalho, V.L.; Pinto da Silva, E.V.; Cardoso, J.F.; Sousa, E.C.; Nunes, K.N.B.; et al. Phylogeography of Dengue Virus Serotype 4, Brazil, 2010–2011. Emerg. Infect. Dis. 2012, 18, 1858–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahadeo, N.S.D.; Allicock, O.M.; De Salazar, P.M.; Auguste, A.J.; Widen, S.; Olowokure, B.; Gutierrez, C.; Valadere, A.M.; Polson-Edwards, K.; Weaver, S.C.; et al. Understanding the Evolution and Spread of Chikungunya Virus in the Americas Using Complete Genome Sequences. Virus Evol. 2017, 3, vex010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Puri, V.; Fedorova, N.; Lin, D.; Hari, K.L.; Jain, R.; Rodas, J.D.; Das, S.R.; Shabman, R.S.; Weaver, S.C. Comprehensive Genome Scale Phylogenetic Study Provides New Insights on the Global Expansion of Chikungunya Virus. J. Virol. 2016, 90, 10600–10611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Severini, F.; Boccolini, D.; Fortuna, C.; Di Luca, M.; Toma, L.; Amendola, A.; Benedetti, E.; Minelli, G.; Romi, R.; Venturi, G.; et al. Vector Competence of Italian Aedes albopictus Populations for the Chikungunya Virus (E1-226V). PLoS Negl. Trop. Dis. 2018, 12, e0006435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsetsarkin, K.A.; Weaver, S.C. Sequential Adaptive Mutations Enhance Efficient Vector Switching by Chikungunya Virus and Its Epidemic Emergence. PLoS Pathog. 2011, 7, e1002412. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Sharma, A.K.; Sukumaran, D.; Parida, M.; Dash, P.K. Two Novel Epistatic Mutations (E1:K211E and E2:V264A) in Structural Proteins of Chikungunya Virus Enhance Fitness in Aedes aegypti. Virology 2016, 497, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Plante, J.A.; Plante, K.S.; Yun, R.; Shinde, D.; Liu, J.; Haller, S.; Mukhopadhyay, S.; Weaver, S.C. Lineage Divergence and Vector-Specific Adaptation Have Driven Chikungunya Virus onto Multiple Adaptive Landscapes. mBio 2021, 12, e02738-21. [Google Scholar] [CrossRef]
- Zouache, K.; Failloux, A.-B. Insect–Pathogen Interactions: Contribution of Viral Adaptation to the Emergence of Vector-Borne Diseases, the Example of Chikungunya. Curr. Opin. Insect Sci. 2015, 10, 14–21. [Google Scholar] [CrossRef]
- Chua, C.-L.; Sam, I.-C.; Chiam, C.-W.; Chan, Y.-F. The Neutralizing Role of IgM during Early Chikungunya Virus Infection. PLoS ONE 2017, 12, e0171989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 Is a Receptor for Multiple Arthritogenic Alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.J.; de Souza, W.M.; Espósito, D.L.A.; Silva, A.; Romeiro, M.F.; Martinez, E.Z.; da Fonseca, B.A.L.; Figueiredo, L.T.M. Enzyme-Linked Immunosorbent Assay Using Recombinant Envelope Protein 2 Antigen for Diagnosis of Chikungunya Virus. Virol. J. 2018, 15, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.D.; Lee, J.E. The Secret Life of Viral Entry Glycoproteins: Moonlighting in Immune Evasion. PLoS Pathog. 2013, 9, e1003258. [Google Scholar] [CrossRef]
- Gräf, T.; Vazquez, C.; Giovanetti, M.; de Bruycker-Nogueira, F.; Fonseca, V.; Claro, I.M.; de Jesus, J.G.; Gómez, A.; Xavier, J.; de Mendonça, M.C.L.; et al. Epidemiologic History and Genetic Diversity Origins of Chikungunya and Dengue Viruses, Paraguay. Emerg. Infect. Dis. 2021, 27, 1393–1404. [Google Scholar] [CrossRef]
- White, S.K.; Mavian, C.; Salemi, M.; Morris, J.G.; Elbadry, M.A.; Okech, B.A.; Lednicky, J.A.; Dunford, J.C. A New “American” Subgroup of African-Lineage Chikungunya Virus Detected in and Isolated from Mosquitoes Collected in Haiti, 2016. PLoS ONE 2018, 13, e0196857. [Google Scholar] [CrossRef] [Green Version]
- De Souza, T.; Ribeiro, E.; Corrêa, V.; Damasco, P.; Santos, C.; de Bruycker-Nogueira, F.; Chouin-Carneiro, T.; Faria, N.; Nunes, P.; Heringer, M.; et al. Following in the Footsteps of the Chikungunya Virus in Brazil: The First Autochthonous Cases in Amapá in 2014 and Its Emergence in Rio de Janeiro during 2016. Viruses 2018, 10, 623. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.C. Arrival of Chikungunya Virus in the New World: Prospects for Spread and Impact on Public Health. PLoS Negl. Trop. Dis. 2014, 8, e2921. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, R.G.; Lourenço-de-Oliveira, R.; Braga, I.A. Updating the Geographical Distribution and Frequency of Aedes albopictus in Brazil with Remarks Regarding Its Range in the Americas. Mem. Inst. Oswaldo Cruz 2014, 109, 787–796. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Collection Date (Y-M-D) | Age 1 | Sex | Municipalities | Cycle Threshold |
|---|---|---|---|---|---|
| 135 | 2014-10-10 | 54 | F | Macapá | 28.21 |
| 139 | 2014-10-24 | 20 | F | Macapá | 22.56 |
| 181 | 2014-10-28 | 29 | F | Macapá | 26.93 |
| 351 | 2015-01-09 | 21 | M | Macapá | 32.77 |
| 539 | 2015-02-03 | 16 | F | Laranjal do Jari | 35.61 |
| 548 | 2015-02-20 | 42 | M | Macapá | 20.64 |
| 564 | 2015-02-03 | 61 | F | Macapá | 37.35 |
| 574 * | 2015-01-29 | 30 | M | Porto Grande | NA |
| 575 | 2015-02-24 | 30 | F | Macapá | 27.93 |
| 613 | 2015-02-25 | 26 | F | Macapá | 20.2 |
| 695 | 2016-03-11 | 28 | M | Macapá | NA |
| Gene | Substitution | Amapá Sequences | Function |
|---|---|---|---|
| E1 | L19M | Yes | Unknow |
| T98A | Yes | Enhanced vector adaptability of A226V | |
| A226V | No | Increased infectivity, transmission, and dissemination in A. Albopictus | |
| E2 | L210Q | No | Enhanced disseminated infection in A. albopictus and fitness increment of A226V variant |
| V226 | Yes * | Unknow | |
| V367A | Yes | Unknow | |
| V384M | Yes | Unknow | |
| nsP1 | R364K | Yes | Unknow |
| Capsid | V54A | Yes | Unknow |
| 6k | L20 | Yes * | Unknow |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira Ribeiro, G.; Gill, D.E.; do Socorro Foro Ramos, E.; Villanova, F.; Soares D’Athaide Ribeiro, E.; Monteiro, F.J.C.; Morais, V.S.; Rego, M.O.d.S.; Araújo, E.L.L.; Pandey, R.P.; et al. Chikungunya Virus Asian Lineage Infection in the Amazon Region Is Maintained by Asiatic and Caribbean-Introduced Variants. Viruses 2022, 14, 1445. https://doi.org/10.3390/v14071445
de Oliveira Ribeiro G, Gill DE, do Socorro Foro Ramos E, Villanova F, Soares D’Athaide Ribeiro E, Monteiro FJC, Morais VS, Rego MOdS, Araújo ELL, Pandey RP, et al. Chikungunya Virus Asian Lineage Infection in the Amazon Region Is Maintained by Asiatic and Caribbean-Introduced Variants. Viruses. 2022; 14(7):1445. https://doi.org/10.3390/v14071445
Chicago/Turabian Stylede Oliveira Ribeiro, Geovani, Danielle Elise Gill, Endrya do Socorro Foro Ramos, Fabiola Villanova, Edcelha Soares D’Athaide Ribeiro, Fred Julio Costa Monteiro, Vanessa S. Morais, Marlisson Octavio da S. Rego, Emerson Luiz Lima Araújo, Ramendra Pati Pandey, and et al. 2022. "Chikungunya Virus Asian Lineage Infection in the Amazon Region Is Maintained by Asiatic and Caribbean-Introduced Variants" Viruses 14, no. 7: 1445. https://doi.org/10.3390/v14071445