
Predictive Models of within- and between-Species SARS-CoV-2 Transmissibility



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval
2.2. Coronaviridae’s Use of ACE2
2.3. Protein–Protein Docking
2.4. S protein: Sites under Positive Selection
2.5. Phylogenetic Inferences
2.6. Large Scale Predictive Model
3. Results
3.1. B Lineage CoVs That Bind to ACE2 Show a Distinctive Amino Acid Pattern
3.2. More Recent SARS-CoV-2 Variants Are Inferred to Have Replaced the Old Ones Because They Are Able to Bind with Higher Affinity to the ACE2 Receptor
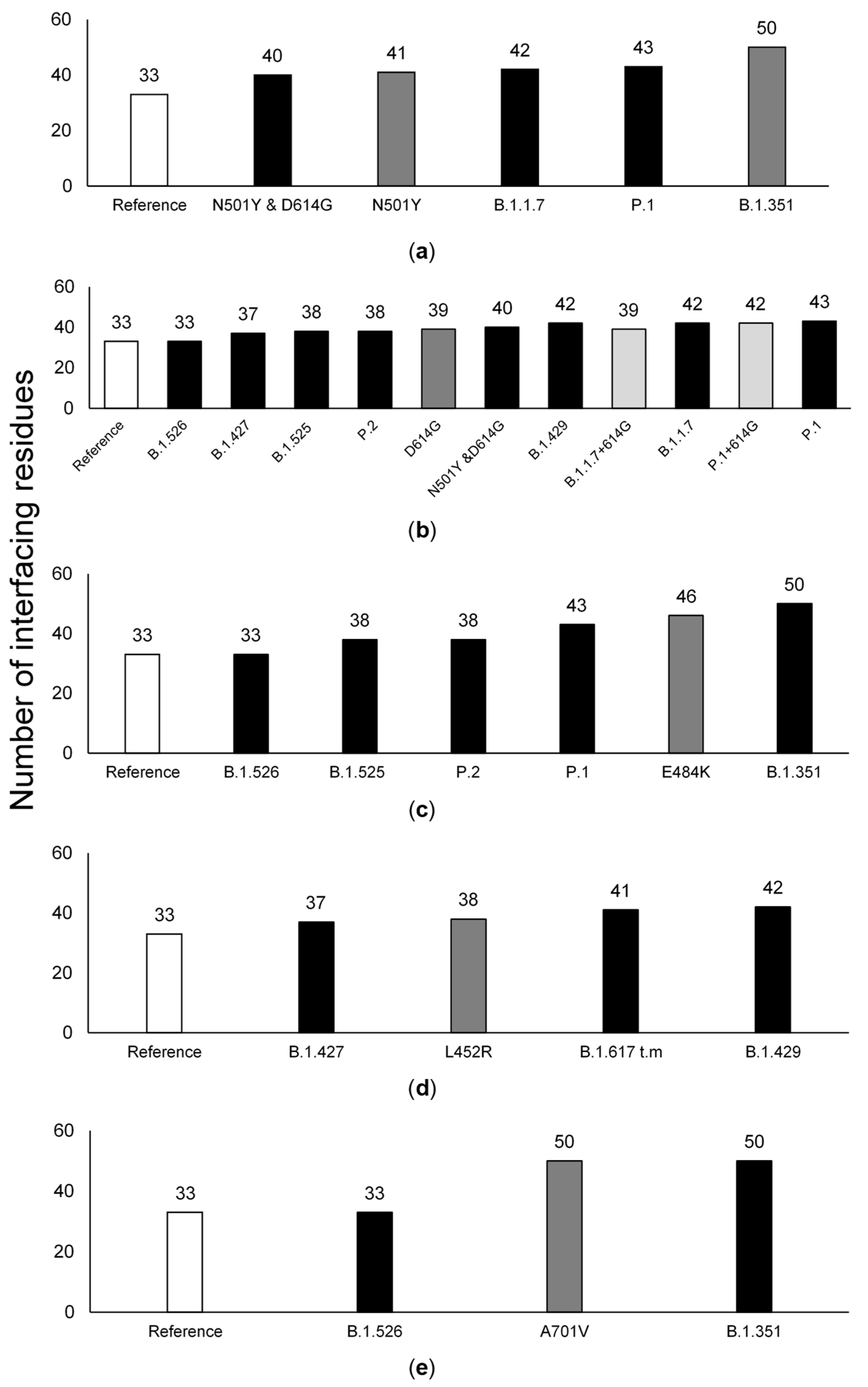
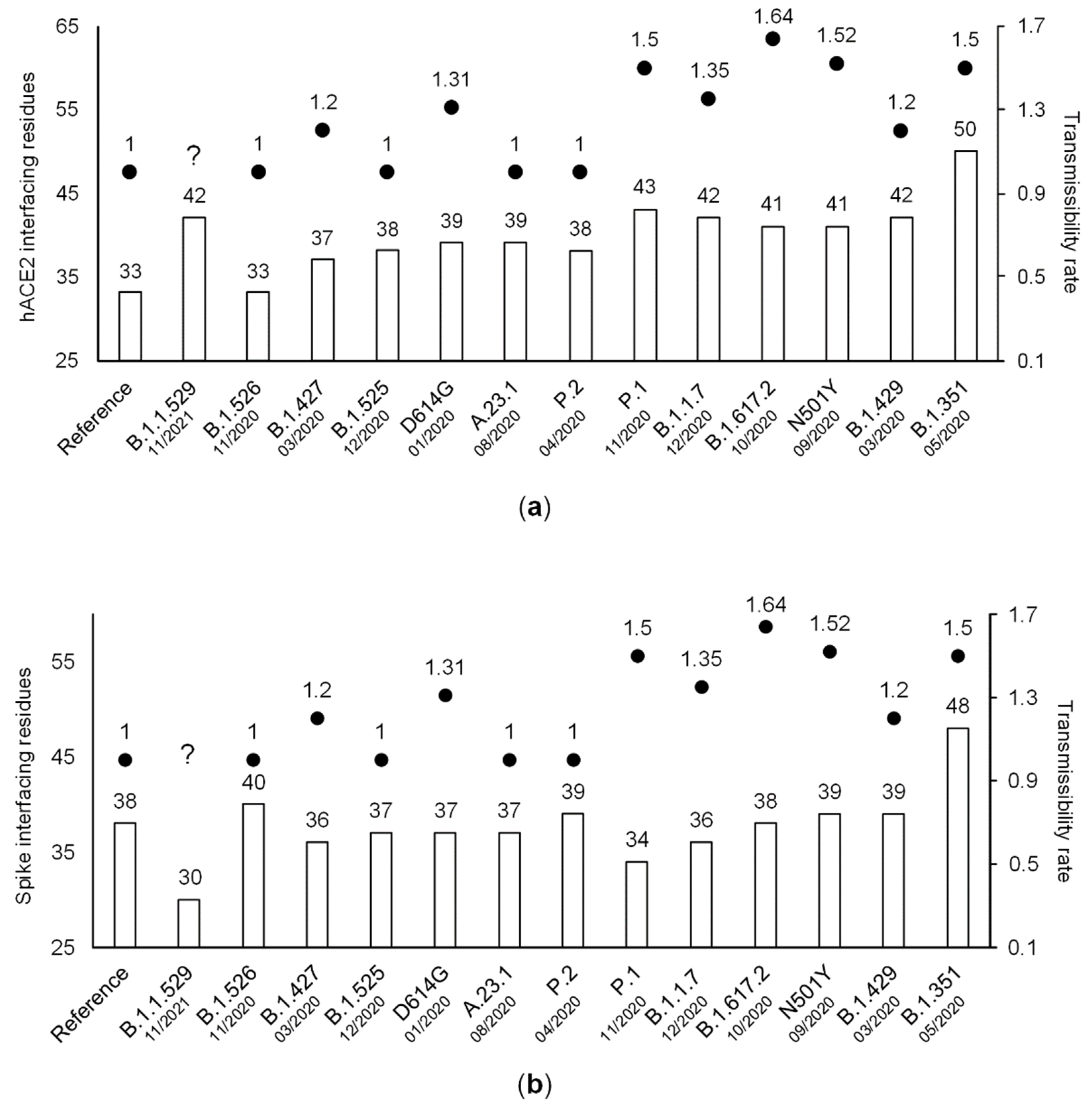
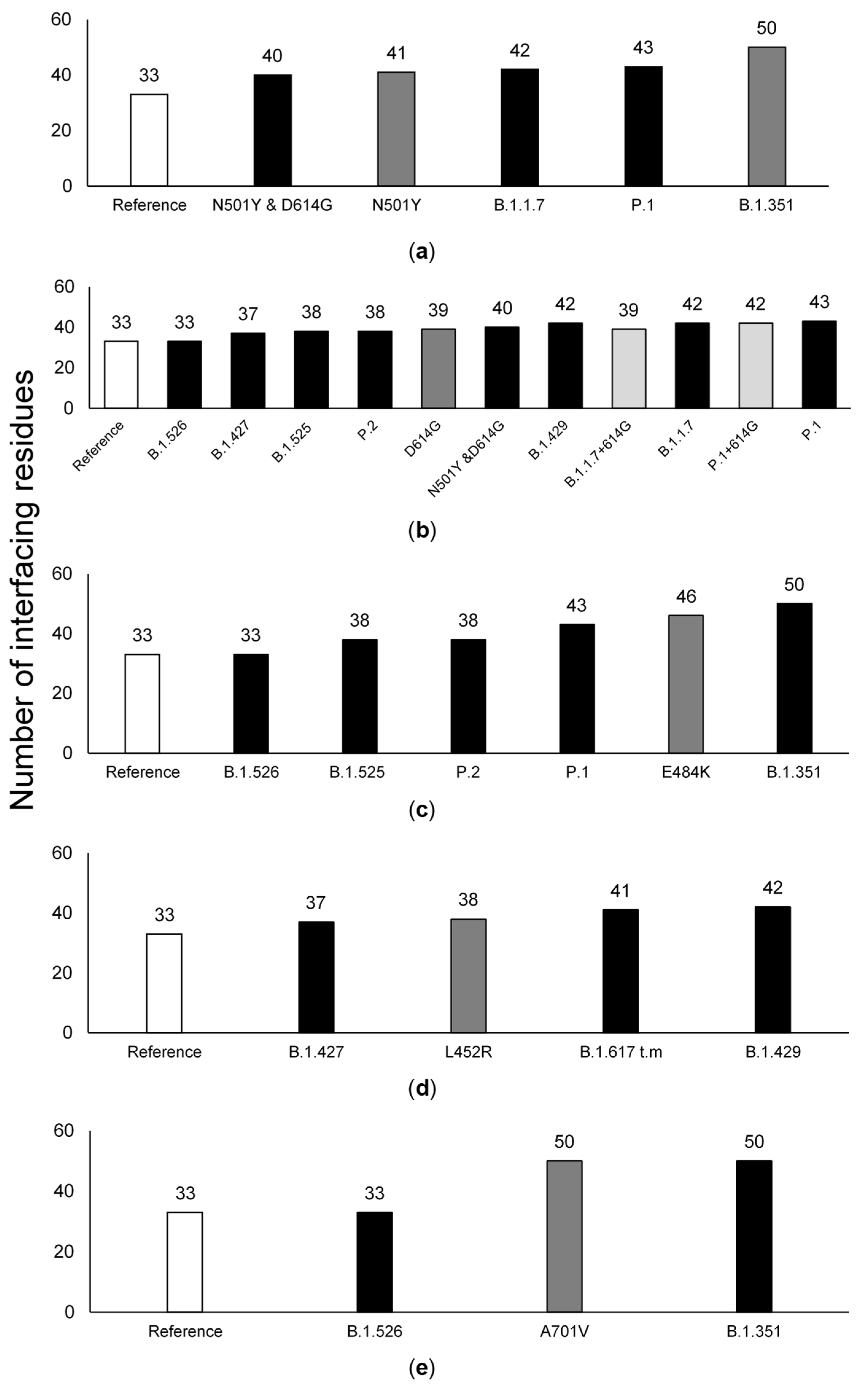
3.3. The Number of ACE2-Interfacing Residues Is Positively Correlated with Transmissibility Rates
3.4. The Effect of Individual Amino Acid Substitutions
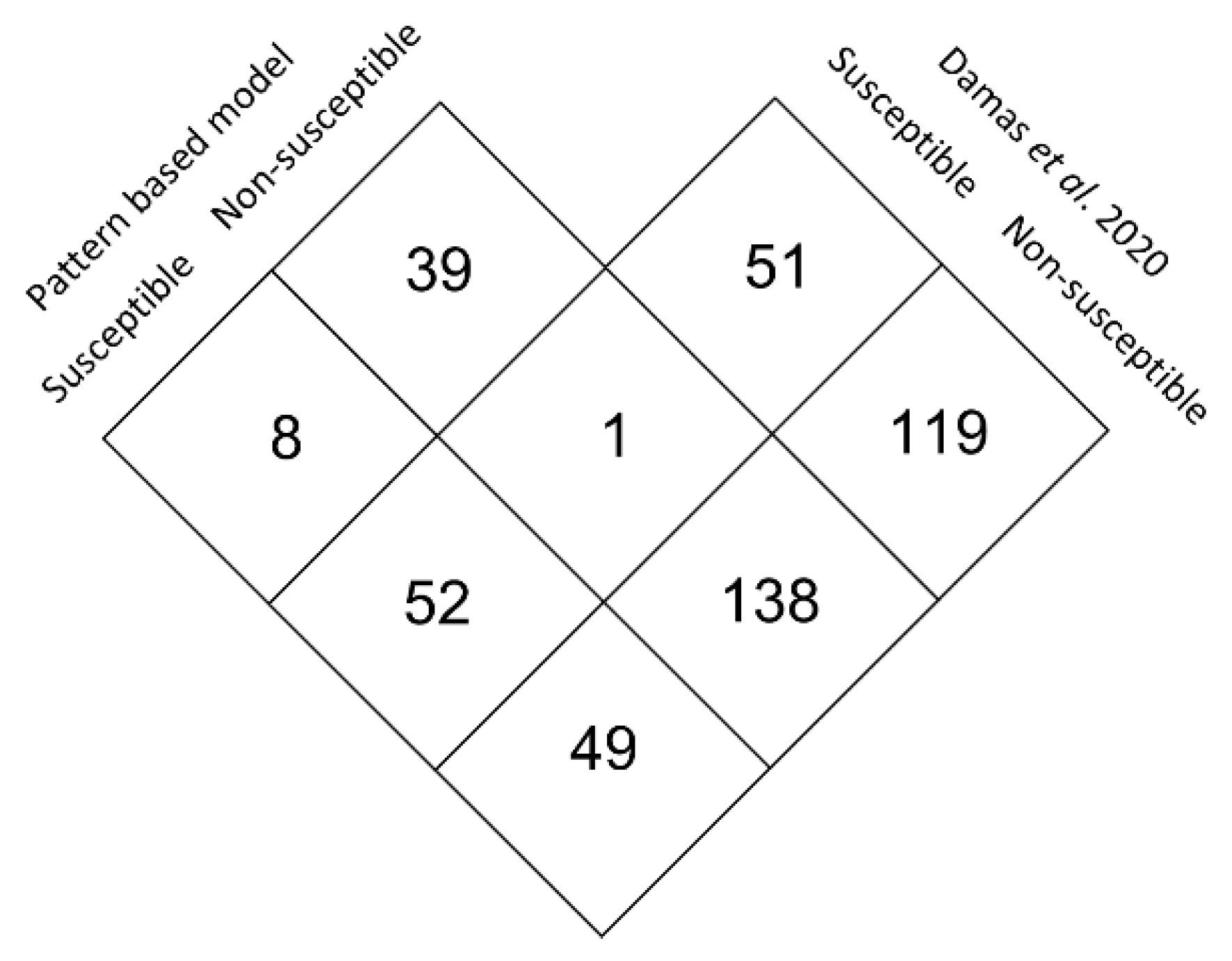
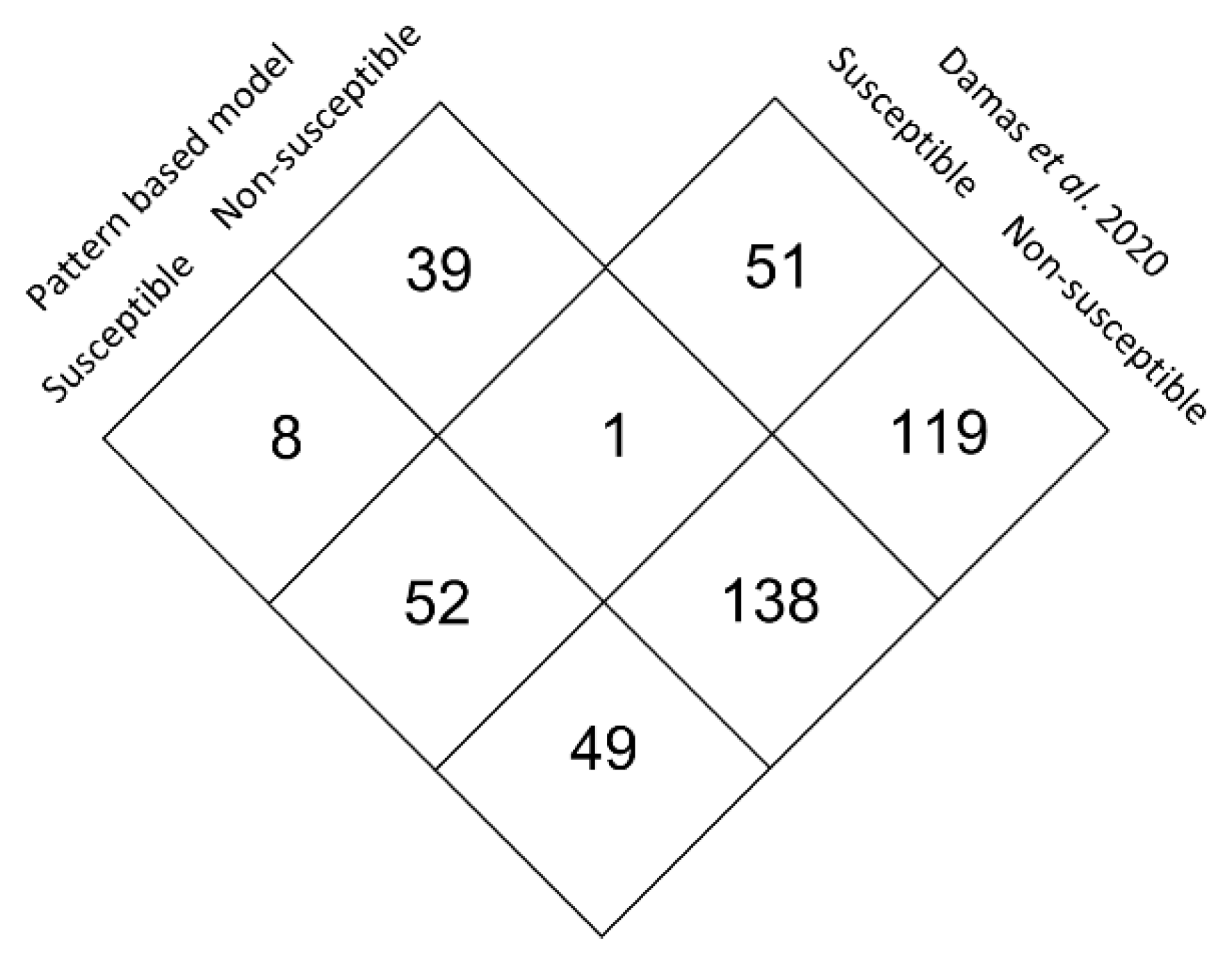
3.5. Large Scale Predictive Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tyrrell, D.; Bynoe, M. Cultivation of a novel type of common-cold virus in organ cultures. BMJ 1965, 1, 1467. [Google Scholar] [CrossRef] [Green Version]
- Millet, J.K.; Jaimes, J.A.; Whittaker, G.R. Molecular diversity of coronavirus host cell entry receptors. FEMS Microbiol. Rev. 2021, 45, fuaa057. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. Severe Acute Respiratory Syndrome, Fact Sheet: Basic Information about SARS. 2004. Available online: https://www.cdc.gov/sars/about/fs-SARS.pd (accessed on 1 November 2021).
- World Health Organization, Regional Offic for the Eastern Mediterranean. MERS Situation Update, January 2020. Available online: https://applications.emro.who.int/docs/EMCSR254E.pdf?ua=1 (accessed on 1 November 2021).
- European Centre for Disease Prevention and Control. COVID-19 Situation Update Worldwide. Available online: https://www.ecdc.europa.eu/en/geographical-distribution-2019-ncov-cases (accessed on 10 January 2021).
- Bosch, B.J.; Van der Zee, R.; De Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2019; Volume 105, pp. 93–116. [Google Scholar]
- Heald-Sargent, T.; Gallagher, T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 2012, 4, 557–580. [Google Scholar] [CrossRef] [Green Version]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Nat. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Walls, A.C.; Xiong, X.; Park, Y.-J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell 2020, 183, 1732. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Carvalho, P.P.; Alves, N.A. Featuring ACE2 binding SARS-CoV and SARS-CoV-2 through a conserved evolutionary pattern of amino acid residues. J. Biomol. Struct. Dyn. 2021, 1–10. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.-P.; Yang, X.-L.; Ge, X.-Y.; Zhang, W.; Li, B.; Xie, J.-Z.; Shen, X.-R.; Zhang, Y.-Z.; Wang, N. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-K.; Chang, W.-C.; Prakash, E.; Peng, Y.-J.; Tu, Z.-J.; Lin, C.-H.; Hsu, P.-H.; Chang, C.-F. Carbohydrate Ligands for COVID-19 Spike Proteins. Viruses 2022, 14, 330. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.E.; Yoo, K.H. Renin-Angiotensin system-considerations for hypertension and kidney. Electrolytes Blood Press. 2008, 6, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Fournier, D.; Luft, F.C.; Bader, M.; Ganten, D.; Andrade-Navarro, M.A. Emergence and evolution of the renin–angiotensin–aldosterone system. J. Mol. Med. 2012, 90, 495–508. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Li, Y.; Yi, Y.; Zhang, L.; Shi, Q.; Yang, J. A genomic survey of angiotensin-converting enzymes provides novel insights into their molecular evolution in vertebrates. Molecules 2018, 23, 2923. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.F. Angiotensin-I-converting enzyme and its relatives. Genome Biol. 2003, 4, 225. [Google Scholar] [CrossRef] [Green Version]
- Warner, F.; Smith, A.; Hooper, N.; Turner, A. Angiotensin-converting enzyme-2: A molecular and cellular perspective. Cell. Mol. Life Sci. CMLS 2004, 61, 2704–2713. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.; Khalid, M.; Servellita, V.; Wang, C.; Morris, M. Transmission, Infectivity, and Antibody Neutralization of an Emerging SARS-CoV-2 Variant in California Carrying a L452R Spike Protein Mutation. In medRxiv; 9 Mars 2021 [Cité 2 Avr 2021]. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7987058 (accessed on 1 November 2021).
- Faria, N.R.; Claro, I.M.; Candido, D.; Franco, L.M.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.; Sales, F.C.; Manuli, E.R.; Aguiar, R.S. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: Preliminary findings. Virological 2021, 372, 815–821. [Google Scholar]
- Graham, M.S.; Sudre, C.H.; May, A.; Antonelli, M.; Murray, B.; Varsavsky, T.; Kläser, K.; Canas, L.S.; Molteni, E.; Modat, M. Changes in symptomatology, reinfection, and transmissibility associated with the SARS-CoV-2 variant B. 1.1. 7: An ecological study. Lancet Public Health 2021, 6, e335–e345. [Google Scholar] [CrossRef]
- Pearson, C.A.B. Estimates of Severity and Transmissibility of Novel South Africa SARS-CoV-2 Variant 501Y.V2. Available online: https://cmmid.github.io/topics/covid19/reports/sa-novel-variant/2021_01_11_Transmissibility_and_severity_of_501Y_V2_in_SA.pdf (accessed on 1 November 2021).
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Zheng, B.; He, Y.; Liu, X.; Zhuang, Z.; Cheung, C.; Luo, S.; Li, P.H.; Zhang, L.; Guan, Y. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Omrani, A.S.; Al-Tawfiq, J.A.; Memish, Z.A. Middle East respiratory syndrome coronavirus (MERS-CoV): Animal to human interaction. Pathog. Global Health 2015, 109, 354–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.-J.; Li, N.; Guo, Y.; Li, X.; Shen, X. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 2020, 583, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.-Y.; Jia, N.; Zhang, Y.-W.; Shum, M.H.-H.; Jiang, J.-F.; Zhu, H.-C.; Tong, Y.-G.; Shi, Y.-X.; Ni, X.-B.; Liao, Y.-S. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [Green Version]
- Damas, J.; Karlsson, E.K.; Lewin, H.A. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Nat. Acad. Sci. USA 2020, 117, 22311–22322. [Google Scholar] [CrossRef]
- Lopez-Fernandez, H.; Duque, P.; Vazquez, N.; Fdez-Riverola, F.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. SEDA: A desktop tool suite for FASTA files processing. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 1850–1860. [Google Scholar] [CrossRef]
- López-Fernández, H.; Ferreira, P.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. The pegi3s Bioinformatics Docker Images Project. In Proceedings of the International Conference on Practical Applications of Computational Biology & Bioinformatics, Salamanca, Spain, 6–8 October 2021; pp. 31–40. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef] [PubMed]
- Gryseels, S.; De Bruyn, L.; Gyselings, R.; Calvignac-Spencer, S.; Leendertz, F.H.; Leirs, H. Risk of human-to-wildlife transmission of SARS-CoV-2. Mamm. Rev. 2021, 51, 272–292. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Chen, Q.; Yang, G.; He, L.; Fan, H.; Deng, Y.-Q.; Wang, Y.; Teng, Y.; Zhao, Z.; Cui, Y. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 2020, 369, 1603–1607. [Google Scholar] [CrossRef]
- Heegaard, P.M.; Sturek, M.; Alloosh, M.; Belsham, G.J. Animal models for COVID-19: More to the picture than ACE2, rodents, ferrets, and non-human primates. A case for porcine respiratory coronavirus and the obese ossabaw pig. Front. Microbiol. 2020, 11, 573756. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; Yeats, C.; et al. Pangolin: Lineage Assignment in an Emerging Pandemic as an Epidemiological Tool. Available online: https://github.com/cov-lineages/pangolin (accessed on 6 May 2021).
- Centers for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variantinfo.html?fbclid=IwAR3YwLYIOMz431yqTo-ZyEiG8V0ruYv_XkrgOZzZUzC77nbROg11bUfm5Qg (accessed on 6 May 2021).
- Gisaid. Available online: https://www.gisaid.org/about-us/acknowledgements/data-curation/ (accessed on 28 January 2022).
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucl. Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; De Vries, S.; Bonvin, A. The HADDOCK2. 2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- López-Fernández, H.; Vieira, C.P.; Ferreira, P.; Gouveia, P.; Fdez-Riverola, F.; Reboiro-Jato, M.; Vieira, J. On the Identification of Clinically Relevant Bacterial Amino Acid Changes at the Whole Genome Level Using Auto-PSS-Genome. Interdiscip. Sci. Comput. Life Sci. 2021, 13, 334–343. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reboiro-Jato, D.; Reboiro-Jato, M.; Fdez-Riverola, F.; Vieira, C.P.; Fonseca, N.A.; Vieira, J. ADOPS-Automatic detection of positively selected sites. J. Int. Bioinform. 2012, 9, 18–32. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingstone, C.D.; Barton, G.J. Protein sequence alignments: A strategy for the hierarchical analysis of residue conservation. Bioinformatics 1993, 9, 745–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhang, Y.; Qu, Y.; Zhang, C.; Liu, X.-W.; Zhao, M.; Mu, Y.; Li, W. Key residues of the receptor binding domain in the spike protein of SARS-CoV-2 mediating the interactions with ACE2: A molecular dynamics study. Nanoscale 2021, 13, 9364–9370. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, W.; Peng, G.; Li, F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Nat. Acad. Sci. USA 2009, 106, 19970–19974. [Google Scholar] [CrossRef] [Green Version]
- Lesté-Lasserre, C. Mutant coronaviruses found in mink spark massive culls and doom a Danish group’s research. Science 2020, 370, 754. [Google Scholar] [CrossRef]
- Meredith, R.W.; Janečka, J.E.; Gatesy, J.; Ryder, O.A.; Fisher, C.A.; Teeling, E.C.; Goodbla, A.; Eizirik, E.; Simão, T.L.; Stadler, T. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science 2011, 334, 521–524. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variants | WHO Label | Mutations |
|---|---|---|
| A.23.1 * | - | F157L; V367F; Q613H; P681R |
| B.1.1.7 * | Alpha | del69–70HV; del144Y; N501Y; A570D; P681H; T716I; S982A; D1118H |
| B.1.351 * | Beta | D80A; D215G; K417N; E484K; N501Y; A701V |
| B.1.427 ** | Epsilon | L452R; D614G |
| B.1.429 ** | Epsilon | S13I; W152C; L452R; D614G |
| B.1.525 ** | Eta | A67V; del69–70 HV; del144 Y; E484K; D614G; Q677H; F888L |
| B.1.526 ** | Iota | L5F; T95I; D253G; S477N; E484K; D614G; A701V |
| P.1 * | Gamma | L18F; T20N; P26S; D138Y; R190S; K417T; E484K; N501Y; H655Y; T1027I |
| P.2 ** | Zeta | E484K; D614G; V1176F |
| B.1.617.2 *** | Delta | T19R; T95I; G142D; E156-; F157-; R158G; L452R; T478K; D614G; P681R; D950N |
| B.1.1.529 *** | Omicron | A67V; H69del; V70del; T95I; G142D; V143del; Y144del; Y145del; N211del; L212I; ins214EPE; G339D; S371L; S373P; S375F; K417N; N440K; G446S; S477N; T478K; E484A; Q493R; G496S; Q498R; N501Y; Y505H; T547K; D614G; H655Y; N679K; P681H; N764K; D796Y; N856K; Q954H; N969K; L981F |
| Method | 25% < PSF 1 < 75% | PSF 1 >75% |
|---|---|---|
| codeML FUBAR | 5, 681, 677 95, 732, 494, 138, 18, 26, 477, 681 | - 5, 484, 501, 677 |
| Protein | Residue Number |
|---|---|
| ACE2 | 30, 31, 34, 35, 37, 38, 41, 42, 353, 354, 386 |
| S | 403, 417, 449, 455, 456, 484, 486, 487, 489, 493, 494, 495, 496, 498, 501, 505 |
| Residue Number | Variation | Common Chemical Characteristic |
|---|---|---|
| 30 | [ADEN] | - |
| 31 | [EKNT] | Polar |
| 34 | [HLQSTVY] | - |
| 35 | [ER] | Big hydrophilic charged polar |
| 37 | [EQ] | Big hydrophilic polar |
| 38 | [DE] | Negatively charged polar |
| 41 | [HY] | Aromatic polar |
| 42 | [EQ] | Big hydrophilic polar |
| 353 | [HK] | Hydrophobic positively charged polar |
| 354 | [DGHNQR] | - |
| 386 | A | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soares, R.; Vieira, C.P.; Vieira, J. Predictive Models of within- and between-Species SARS-CoV-2 Transmissibility. Viruses 2022, 14, 1565. https://doi.org/10.3390/v14071565
Soares R, Vieira CP, Vieira J. Predictive Models of within- and between-Species SARS-CoV-2 Transmissibility. Viruses. 2022; 14(7):1565. https://doi.org/10.3390/v14071565
Chicago/Turabian StyleSoares, Ricardo, Cristina P. Vieira, and Jorge Vieira. 2022. "Predictive Models of within- and between-Species SARS-CoV-2 Transmissibility" Viruses 14, no. 7: 1565. https://doi.org/10.3390/v14071565