
Implications of the Actin Cytoskeleton on the Multi-Step Process of [PSI+] Prion Formation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Cultivation Procedures
2.2. Sup35NM Early Foci and Aggregate Microscopy
2.3. 3D Time-Lapse Microscopy and Coordinate Mapping
2.4. Western Blot Analysis
2.5. De Novo [PSI+] Induction
3. Results
3.1. Co-Localization between Sup35NM-GFP Early Foci and Actin Networks Is Inconclusive
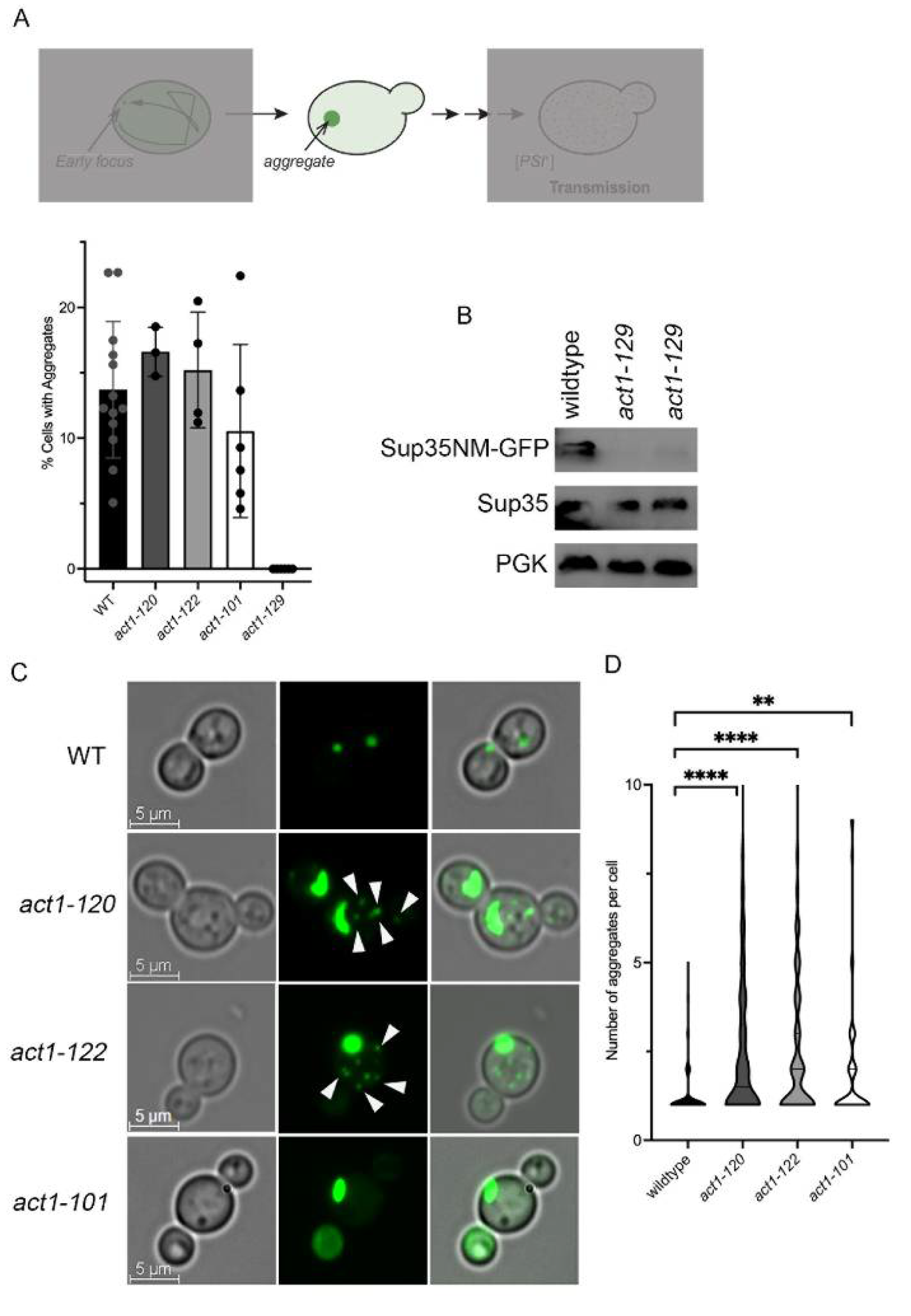
3.2. Actin Polarization Mutants form Multiple Sup35NM-GFP Aggregates
3.3. Early Foci Size Is Correlated with Movement Variability, but Early Foci Movement Is Random
3.4. Sup35NM Aggregates Do Not Localize with Organelles or IPOD
3.5. Hsp104 Co-Localizes to Early Foci Intermittently
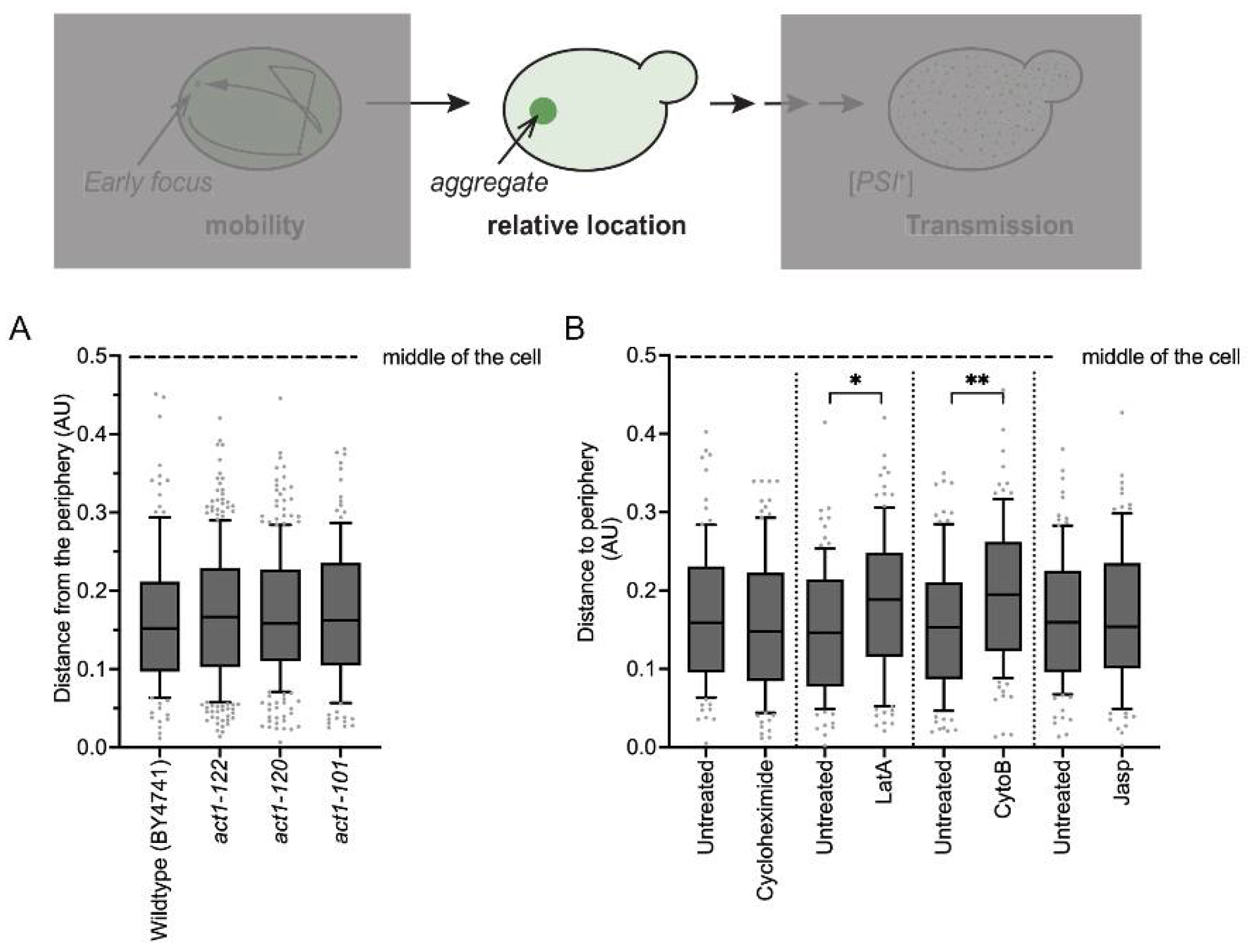
3.6. Actin Fibril Disruption Changes Aggregate Position
3.7. act1-122 and act1-101 Significantly Alter Prion Induction Frequency
4. Discussion
4.1. Mobility of Early Foci and Localization
4.2. Actin Networks and Aggregate Coalescence
4.3. Actin and the Transmission of Propagons
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. Embo J. 1996, 15, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J.; Lindquist, S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 2004, 304, 1793–1797. [Google Scholar] [CrossRef] [Green Version]
- Satpute-Krishnan, P.; Langseth, S.X.; Serio, T.R. Hsp104-dependent remodeling of prion complexes mediates protein-only inheritance. PLoS Biol. 2007, 5, e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aigle, M.; Lacroute, F. Genetical aspects of [URE3], a non-mitochondrial, cytoplasmically inherited mutation in yeast. Mol. Gen. Genet. 1975, 136, 327–335. [Google Scholar] [CrossRef]
- Allen, K.D.; Chernova, T.A.; Tennant, E.P.; Wilkinson, K.D.; Chernoff, Y.O. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J. Biol. Chem. 2007, 282, 3004–3013. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, A.K.; Bardill, J.P.; True, H.L.; Masel, J. The spontaneous appearance rate of the yeast prion [PSI+] and its implications for the evolution of the evolvability properties of the [PSI+] system. Genetics 2010, 184, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Chernoff, Y.O.; Derkach, I.L.; Inge-Vechtomov, S.G. Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr. Genet. 1993, 24, 268–270. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Chernoff, Y.O.; Kushnirov, V.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996, 144, 1375–1386. [Google Scholar] [CrossRef]
- Wickner, R.B. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN(+)]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Keefer, K.M.; Stein, K.C.; True, H.L. Heterologous prion-forming proteins interact to cross-seed aggregation in Saccharomyces cerevisiae. Sci. Rep. 2017, 7, 5853. [Google Scholar] [CrossRef] [PubMed]
- Villali, J.; Dark, J.; Brechtel, T.M.; Pei, F.; Sindi, S.S.; Serio, T.R. Nucleation seed size determines amyloid clearance and establishes a barrier to prion appearance in yeast. Nat. Struct. Mol. Biol. 2020, 27, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Derkatch, I.L.; Liebman, S.W. The relationship between visible intracellular aggregates that appear after overexpression of Sup35 and the yeast prion-like elements [PSI(+)] and [PIN(+)]. Mol. Microbiol. 2001, 39, 37–46. [Google Scholar] [CrossRef]
- Sharma, J.; Wisniewski, B.T.; Paulson, E.; Obaoye, J.O.; Merrill, S.J.; Manogaran, A.L. De novo [PSI +] prion formation involves multiple pathways to form infectious oligomers. Sci. Rep. 2017, 7, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, V.; Taneja, V.; Sun, Y.; Liebman, S.W. Analyzing the birth and propagation of two distinct prions, [PSI+] and [Het-s](y), in yeast. Mol. Biol. Cell 2010, 21, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Arslan, F.; Hong, J.Y.; Kanneganti, V.; Park, S.K.; Liebman, S.W. Heterologous aggregates promote de novo prion appearance via more than one mechanism. PLoS Genet. 2015, 11, e1004814. [Google Scholar] [CrossRef] [Green Version]
- Ganusova, E.E.; Ozolins, L.N.; Bhagat, S.; Newnam, G.P.; Wegrzyn, R.D.; Sherman, M.Y.; Chernoff, Y.O. Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol. Cell. Biol. 2006, 26, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Manogaran, A.L.; Hong, J.Y.; Hufana, J.; Tyedmers, J.; Lindquist, S.; Liebman, S.W. Prion formation and polyglutamine aggregation are controlled by two classes of genes. PLoS Genet. 2011, 7, e1001386. [Google Scholar] [CrossRef] [Green Version]
- Bailleul, P.A.; Newnam, G.P.; Steenbergen, J.N.; Chernoff, Y.O. Genetic study of interactions between the cytoskeletal assembly protein sla1 and prion-forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae. Genetics 1999, 153, 81–94. [Google Scholar] [CrossRef]
- Speldewinde, S.H.; Doronina, V.A.; Tuite, M.F.; Grant, C.M. Disrupting the cortical actin cytoskeleton points to two distinct mechanisms of yeast [PSI+] prion formation. PLoS Genet. 2017, 13, e1006708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernova, T.A.; Kiktev, D.A.; Romanyuk, A.V.; Shanks, J.R.; Laur, O.; Ali, M.; Ghosh, A.; Kim, D.; Yang, Z.; Mang, M.; et al. Yeast Short-Lived Actin-Associated Protein Forms a Metastable Prion in Response to Thermal Stress. Cell Rep. 2017, 18, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Sherman, F.; Fink, G.R.; Hicks, J.B. Methods in Yeast Genetics; Cold Spring Harbor Lab.: Plainview, NY, USA, 1986. [Google Scholar]
- Jonsen, I.D.; Flemming, J.M.; Myers, R.A. Robust state-space modeling of animal movement data. Ecology 2005, 86, 2874–2880. [Google Scholar] [CrossRef]
- Jonsen, I.D.; Myers, R.A.; Flemming, J.M. Meta-analysis of animal movement using state-space models. Ecology 2003, 84, 3055–3063. [Google Scholar] [CrossRef]
- Manogaran, A.L.; Kirkland, K.T.; Liebman, S.W. An engineered nonsense URA3 allele provides a versatile system to detect the presence, absence and appearance of the [PSI+] prion in Saccharomyces cerevisiae. Yeast 2006, 23, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.C.; Galletta, B.J.; Sept, D.; Cooper, J.A. Overlapping and distinct functions for cofilin, coronin and Aip1 in actin dynamics in vivo. J. Cell Sci. 2010, 123, 1329–1342. [Google Scholar] [CrossRef] [Green Version]
- Asakura, T.; Sasaki, T.; Nagano, F.; Satoh, A.; Obaishi, H.; Nishioka, H.; Imamura, H.; Hotta, K.; Tanaka, K.; Nakanishi, H.; et al. Isolation and characterization of a novel actin filament-binding protein from Saccharomyces cerevisiae. Oncogene 1998, 16, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.C.; Pon, L.A. Actin cable dynamics in budding yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, B.T.; Sharma, J.; Legan, E.R.; Paulson, E.; Merrill, S.J.; Manogaran, A.L. Toxicity and infectivity: Insights from de novo prion formation. Curr. Genet. 2017, 64, 117–123. [Google Scholar] [CrossRef]
- Dorweiler, J.E.; Oddo, M.J.; Lyke, D.R.; Reilly, J.A.; Wisniewski, B.T.; Davis, E.E.; Kuborn, A.M.; Merrill, S.J.; Manogaran, A.L. The actin cytoskeletal network plays a role in yeast prion transmission and contributes to prion stability. Mol. Microbiol. 2020, 114, 480–494. [Google Scholar] [CrossRef]
- Wertman, K.F.; Drubin, D.G.; Botstein, D. Systematic mutational analysis of the yeast ACT1 gene. Genetics 1992, 132, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Mathur, V.; Hong, J.Y.; Liebman, S.W. Ssa1 Overexpression and [PIN(+)] Variants Cure [PSI(+)] by Dilution of Aggregates. J. Mol. Biol. 2009, 390, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Tyedmers, J.; Treusch, S.; Dong, J.; McCaffery, J.M.; Bevis, B.; Lindquist, S. Prion induction involves an ancient system for the sequestration of aggregated proteins and heritable changes in prion fragmentation. Proc. Natl. Acad. Sci. USA 2010, 107, 8633–8638. [Google Scholar] [CrossRef] [Green Version]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Rothe, S.; Prakash, A.; Tyedmers, J. The Insoluble Protein Deposit (IPOD) in Yeast. Front. Mol. Neurosci. 2018, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Specht, S.; Miller, S.B.; Mogk, A.; Bukau, B. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J. Cell Biol. 2011, 195, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Escusa-Toret, S.; Vonk, W.I.; Frydman, J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol. 2013, 15, 1231–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grousl, T.; Ungelenk, S.; Miller, S.; Ho, C.T.; Khokhrina, M.; Mayer, M.P.; Bukau, B.; Mogk, A. A prion-like domain in Hsp42 drives chaperone-facilitated aggregation of misfolded proteins. J. Cell Biol. 2018, 217, 1269–1285. [Google Scholar] [CrossRef]
- Drubin, D.G.; Jones, H.D.; Wertman, K.F. Actin structure and function: Roles in mitochondrial organization and morphogenesis in budding yeast and identification of the phalloidin-binding site. Mol. Biol. Cell 1993, 4, 1277–1294. [Google Scholar] [CrossRef]
- Davis, C.M.; Gruebele, M. Cytoskeletal Drugs Modulate Off-Target Protein Folding Landscapes Inside Cells. Biochemistry 2020, 59, 2650–2659. [Google Scholar] [CrossRef]
- Lyke, D.R.; Manogaran, A.L. Spatial sequestration and oligomer remodeling during de novo [PSI+] formation. Prion 2017, 11, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Nawroth, P.P.; Tyedmers, J. Prion Aggregates Are Recruited to the Insoluble Protein Deposit (IPOD) via Myosin 2-Based Vesicular Transport. PLoS Genet. 2016, 12, e1006324. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Liebman, S.W. [PSI(+) ] prion variant establishment in yeast. Mol. Microbiol. 2012, 86, 866–881. [Google Scholar] [CrossRef] [Green Version]
- Derdowski, A.; Sindi, S.S.; Klaips, C.L.; DiSalvo, S.; Serio, T.R. A size threshold limits prion transmission and establishes phenotypic diversity. Science 2010, 330, 680–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erjavec, N.; Nystrom, T. Sir2p-dependent protein segregation gives rise to a superior reactive oxygen species management in the progeny of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2007, 104, 10877–10881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessarz, P.; Schwarz, M.; Mogk, A.; Bukau, B. The yeast AAA+ chaperone Hsp104 is part of a network that links the actin cytoskeleton with the inheritance of damaged proteins. Mol. Cell Biol. 2009, 29, 3738–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Larsson, L.; Franssens, V.; Hao, X.; Hill, S.M.; Andersson, V.; Hoglund, D.; Song, J.; Yang, X.; Oling, D.; et al. Segregation of protein aggregates involves actin and the polarity machinery. Cell 2011, 147, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Yang, Q.; Yang, J.; Larsson, L.; Hao, X.; Zhu, X.; Malmgren-Hill, S.; Cvijovic, M.; Fernandez-Rodriguez, J.; Grantham, J.; et al. Essential genetic interactors of SIR2 required for spatial sequestration and asymmetrical inheritance of protein aggregates. PLoS Genet. 2014, 10, e1004539. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Zhou, P.; Chernoff, Y.O.; Liebman, S.W. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997, 147, 507–519. [Google Scholar] [CrossRef]
- Bradley, M.E.; Edskes, H.K.; Hong, J.Y.; Wickner, R.B.; Liebman, S.W. Interactions among prions and prion "strains" in yeast. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. 4), 16392–16399. [Google Scholar] [CrossRef] [Green Version]
- Huh, W.K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Hochstrasser, M.L.; Langlois, C.R.; Serio, T.R. Spatial quality control bypasses cell-based limitations on proteostasis to promote prion curing. eLife 2014, 3, e04288. [Google Scholar] [CrossRef] [PubMed]
- Sondheimer, N.; Lindquist, S. Rnq1: An epigenetic modifier of protein function in yeast. Mol. Cell 2000, 5, 163–172. [Google Scholar] [CrossRef]
- Buttery, S.M.; Yoshida, S.; Pellman, D. Yeast formins Bni1 and Bnr1 utilize different modes of cortical interaction during the assembly of actin cables. Mol. Biol. Cell 2007, 18, 1826–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Valtierra, S.; Li, L. An insight into the complex prion-prion interaction network in the budding yeast Saccharomyces cerevisiae. Prion 2014, 8, 387–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.W.; Miao, Y.H.; Chang, Y.S. Control of lipid droplet size in budding yeast requires the collaboration between Fld1 and Ldb16. J. Cell Sci. 2014, 127, 1214–1228. [Google Scholar] [CrossRef] [Green Version]
- Naylor, K.; Ingerman, E.; Okreglak, V.; Marino, M.; Hinshaw, J.E.; Nunnari, J. Mdv1 interacts with assembled dnm1 to promote mitochondrial division. J. Biol. Chem. 2006, 281, 2177–2183. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Yang, Z.; Liu, X.; Mao, K.; Nair, U.; Klionsky, D.J. Phosphatidylinositol 4-kinases are required for autophagic membrane trafficking. J. Biol. Chem. 2012, 287, 37964–37972. [Google Scholar] [CrossRef] [Green Version]
- Malinovska, L.; Kroschwald, S.; Munder, M.C.; Richter, D.; Alberti, S. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spatiotemporal distribution of protein aggregates. Mol. Biol. Cell 2012, 23, 3041–3056. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorweiler, J.E.; Lyke, D.R.; Lemoine, N.P.; Guereca, S.; Buchholz, H.E.; Legan, E.R.; Radtke, C.M.; Manogaran, A.L. Implications of the Actin Cytoskeleton on the Multi-Step Process of [PSI+] Prion Formation. Viruses 2022, 14, 1581. https://doi.org/10.3390/v14071581
Dorweiler JE, Lyke DR, Lemoine NP, Guereca S, Buchholz HE, Legan ER, Radtke CM, Manogaran AL. Implications of the Actin Cytoskeleton on the Multi-Step Process of [PSI+] Prion Formation. Viruses. 2022; 14(7):1581. https://doi.org/10.3390/v14071581
Chicago/Turabian StyleDorweiler, Jane E., Douglas R. Lyke, Nathan P. Lemoine, Samantha Guereca, Hannah E. Buchholz, Emily R. Legan, Claire M. Radtke, and Anita L. Manogaran. 2022. "Implications of the Actin Cytoskeleton on the Multi-Step Process of [PSI+] Prion Formation" Viruses 14, no. 7: 1581. https://doi.org/10.3390/v14071581