

Analysis of Pseudomonas aeruginosa Isolates from Patients with Cystic Fibrosis Revealed Novel Groups of Filamentous Bacteriophages


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Bacterial Genome Sequencing and Annotation
2.2. Search for Integrated Filamentous Prophages
2.3. Prophage Genomic Analysis
3. Results
3.1. Collection of Genomic Data of Known Pf Phages
3.2. General Characterisation of Pf prophages Found in Genomic Sequences of P. aeruginosa Present in Clinical Isolates

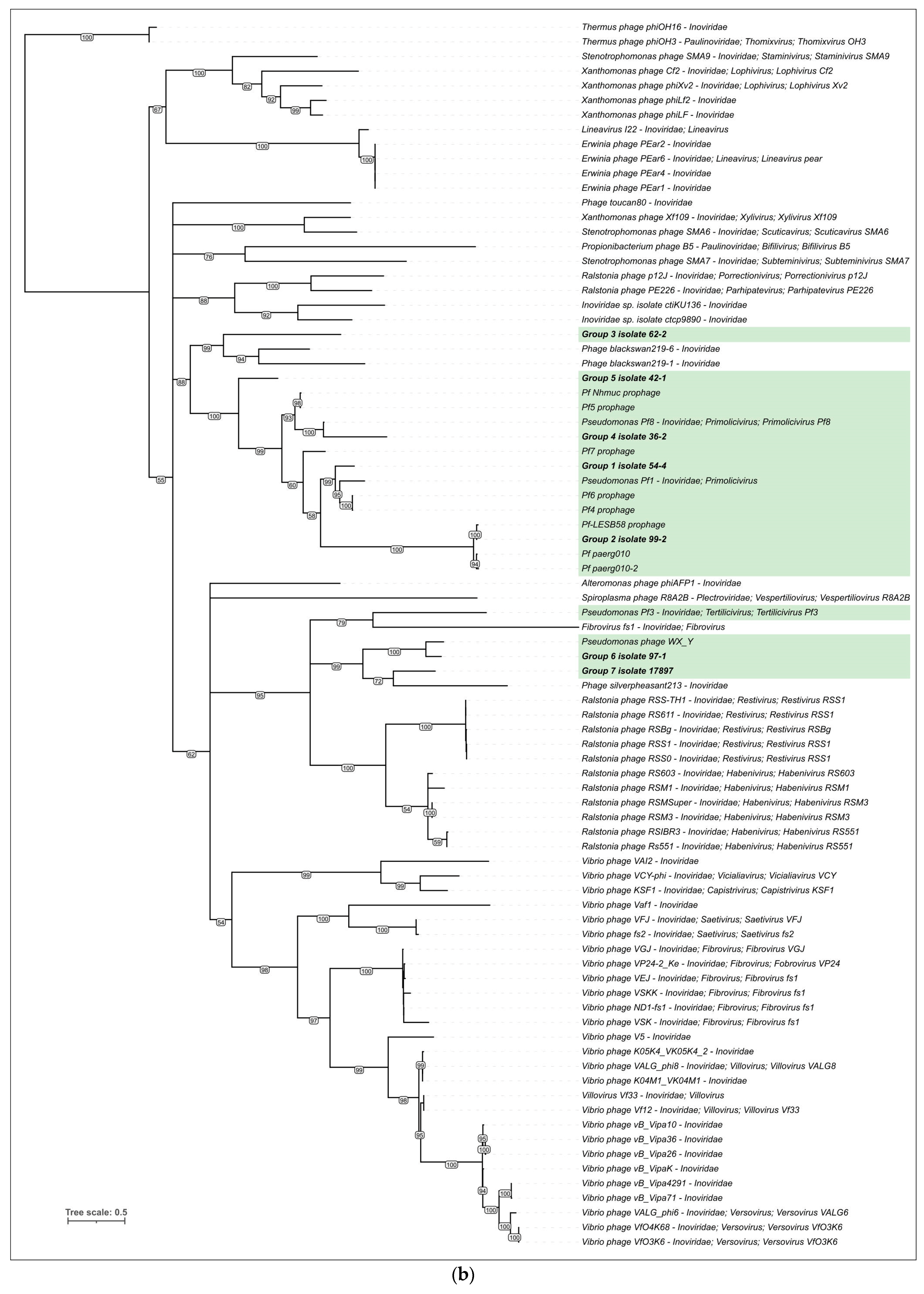
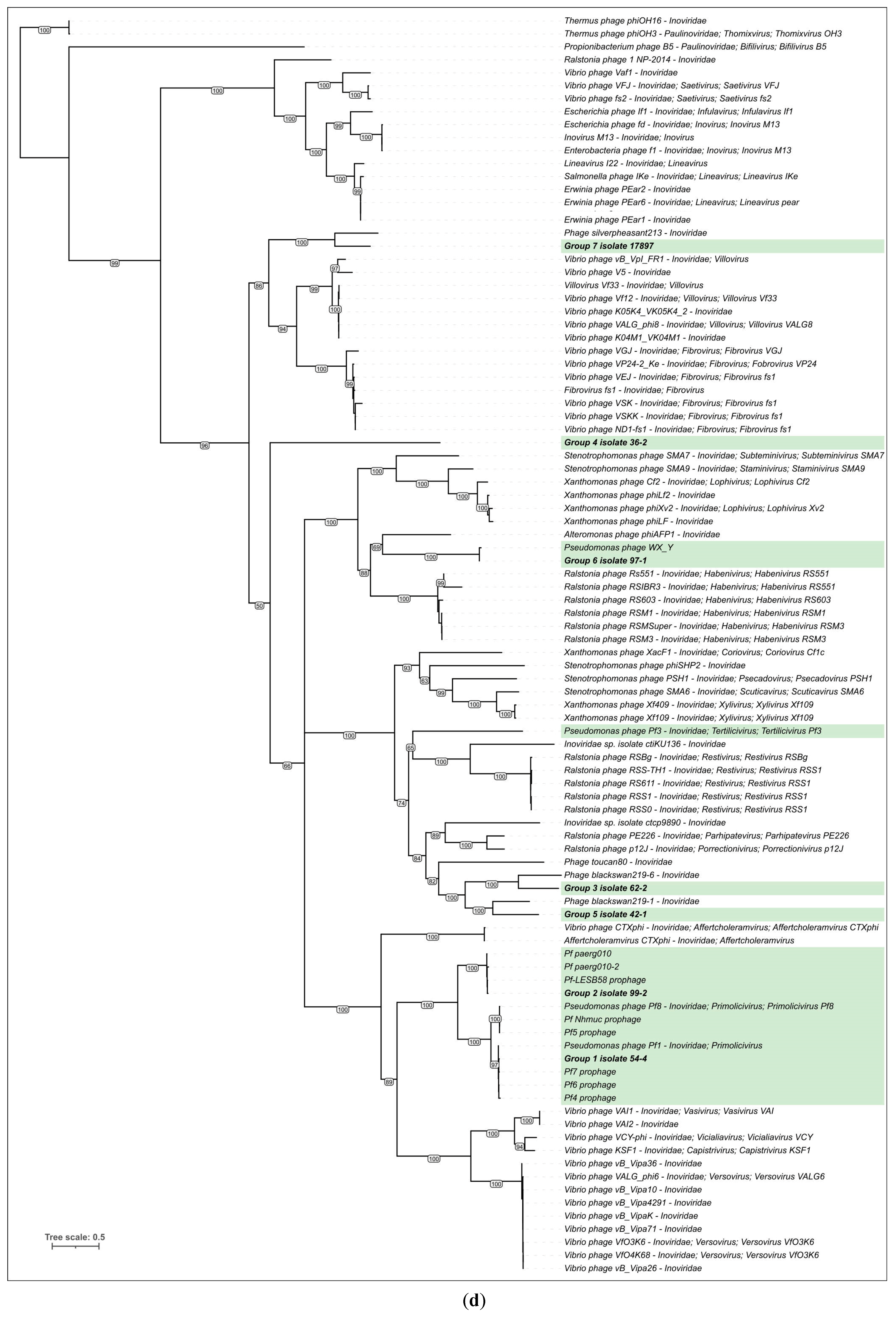
3.3. Intergenomic Comparisons and Phylogenetic Analysis
3.3.1. Genome Alignment
3.3.2. VIRIDIC Analysis
3.3.3. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boucher, R.C. New Concepts of the Pathogenesis of Cystic Fibrosis Lung Disease. Eur. Respir. J. 2004, 23, 146–158. [Google Scholar] [CrossRef]
- Polgreen, P.M.; Comellas, A.P. Clinical Phenotypes of Cystic Fibrosis Carriers. Annu. Rev. Med. 2022, 73, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Annual Reports. European Cystic Fibrosis Society (ECFS). Available online: https://www.ecfs.eu/projects/ecfs-patient-registry/annual-reports (accessed on 5 October 2023).
- Chung, J.; Eisha, S.; Park, S.; Morris, A.J.; Martin, I. How Three Self-Secreted Biofilm Exopolysaccharides of Pseudomonas Aeruginosa, Psl, Pel, and Alginate, Can Each Be Exploited for Antibiotic Adjuvant Effects in Cystic Fibrosis Lung Infection. Int. J. Mol. Sci. 2023, 24, 8709. [Google Scholar] [CrossRef]
- Secor, P.R.; Burgener, E.B.; Kinnersley, M.; Jennings, L.K.; Roman-Cruz, V.; Popescu, M.; Van Belleghem, J.D.; Haddock, N.; Copeland, C.; Michaels, L.A.; et al. Pf Bacteriophage and Their Impact on Pseudomonas Virulence, Mammalian Immunity, and Chronic Infections. Front. Immunol. 2020, 11, 244. [Google Scholar] [CrossRef]
- Hay, I.D.; Lithgow, T. Filamentous Phages: Masters of a Microbial Sharing Economy. EMBO Rep. 2019, 20, e47427. [Google Scholar] [CrossRef]
- Mai-Prochnow, A.; Hui, J.G.K.; Kjelleberg, S.; Rakonjac, J.; McDougald, D.; Rice, S.A. Big Things in Small Packages: The Genetics of Filamentous Phage and Effects on Fitness of Their Host. FEMS Microbiol. Rev. 2015, 39, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Fiedoruk, K.; Zakrzewska, M.; Daniluk, T.; Piktel, E.; Chmielewska, S.; Bucki, R. Two Lineages of Pseudomonas Aeruginosa Filamentous Phages: Structural Uniformity over Integration Preferences. Genome Biol. Evol. 2020, 12, 1765–1781. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, P.; Voet, M.; Lavigne, R. Prevalence of Pf1-like (pro)Phage Genetic Elements among Pseudomonas Aeruginosa Isolates. Virology 2015, 483, 64–71. [Google Scholar] [CrossRef]
- Burgener, E.B.; Sweere, J.M.; Bach, M.S.; Secor, P.R.; Haddock, N.; Jennings, L.K.; Marvig, R.L.; Johansen, H.K.; Rossi, E.; Cao, X.; et al. Filamentous Bacteriophages Are Associated with Chronic Pseudomonas Lung Infections and Antibiotic Resistance in Cystic Fibrosis. Sci. Transl. Med. 2019, 11, eaau9748. [Google Scholar] [CrossRef]
- James, C.E.; Fothergill, J.L.; Kade, H.; Hall, A.J.; Cottell, J.; Brockhurst, M.A.; Winstanley, C. Differential Infection Properties of Three Inducible Prophages from an Epidemic Strain of Pseudomonas Aeruginosa. BMC Microbiol. 2012, 12, 216. [Google Scholar] [CrossRef]
- Gavric, D.; Knezevic, P. Filamentous Pseudomonas Phage Pf4 in the Context of Therapy-Inducibility, Infectivity, Lysogenic Conversion, and Potential Application. Viruses 2022, 14, 1261. [Google Scholar] [CrossRef] [PubMed]
- Winstanley, C.; Langille, M.G.I.; Fothergill, J.L.; Kukavica-Ibrulj, I.; Paradis-Bleau, C.; Sanschagrin, F.; Thomson, N.R.; Winsor, G.L.; Quail, M.A.; Lennard, N.; et al. Newly Introduced Genomic Prophage Islands Are Critical Determinants of in Vivo Competitiveness in the Liverpool Epidemic Strain of Pseudomonas Aeruginosa. Genome Res. 2009, 19, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Gaïa, M.; Meng, L.; Pelletier, E.; Forterre, P.; Vanni, C.; Fernandez-Guerra, A.; Jaillon, O.; Wincker, P.; Ogata, H.; Krupovic, M.; et al. Mirusviruses Link Herpesviruses to Giant Viruses. Nature 2023, 616, 783–789. [Google Scholar] [CrossRef]
- Skewes-Cox, P.; Sharpton, T.J.; Pollard, K.S.; DeRisi, J.L. Profile Hidden Markov Models for the Detection of Viruses within Metagenomic Sequence Data. PLoS ONE 2014, 9, e105067. [Google Scholar] [CrossRef] [PubMed]
- Bocharova, Y.; Chebotar, I.; Savinova, T.; Lyamin, A.; Kondratenko, O.; Polikarpova, S.; Fedorova, N.; Semykin, S.; Korostin, D.; Chaplin, A.; et al. Clonal Diversity, Antimicrobial Resistance, and Genome Features among Non-Fermenting Gram-Negative Bacteria Isolated from Patients with Cystic Fibrosis in Russia. Diagn. Microbiol. Infect. Dis. 2023, in press. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinforma. Oxf. Engl. 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinforma. Oxf. Engl. 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-Suite3 for Fast Remote Homology Detection and Deep Protein Annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap.Js: Automatic Generation of Gene Cluster Comparison Figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Uzzau, S.; Cappuccinelli, P.; Fasano, A. Expression of Vibrio Cholerae Zonula Occludens Toxin and Analysis of Its Subcellular Localization. Microb. Pathog. 1999, 27, 377–385. [Google Scholar] [CrossRef]
- Kedem, S.; Hassid, R.R.; Shamir, Y.; Goldbourt, A. Conformational Changes in Ff Phage Protein gVp upon Complexation with Its Viral Single-Stranded DNA Revealed Using Magic-Angle Spinning Solid-State NMR. Viruses 2022, 14, 1264. [Google Scholar] [CrossRef]
- Wang, W.; Li, Y.; Tang, K.; Lin, J.; Gao, X.; Guo, Y.; Wang, X. Filamentous Prophage Capsid Proteins Contribute to Superinfection Exclusion and Phage Defence in Pseudomonas Aeruginosa. Environ. Microbiol. 2022, 24, 4285–4298. [Google Scholar] [CrossRef]
- Sartorius, R.; D’Apice, L.; Prisco, A.; De Berardinis, P. Arming Filamentous Bacteriophage, a Nature-Made Nanoparticle, for New Vaccine and Immunotherapeutic Strategies. Pharmaceutics 2019, 11, 437. [Google Scholar] [CrossRef]
- McLeod, S.M.; Kimsey, H.H.; Davis, B.M.; Waldor, M.K. CTXφ and Vibrio Cholerae: Exploring a Newly Recognized Type of Phage–Host Cell Relationship. Mol. Microbiol. 2005, 57, 347–356. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, G.; Zhong, L.; Qian, M.; Wang, M.; Cui, R. Filamentous Bacteriophages, Natural Nanoparticles, for Viral Vaccine Strategies. Nanoscale 2022, 14, 5942–5959. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A. Phage Evolution and Speciation. In The Bacteriophages; Calendar, R., Ed.; The Viruses; Springer: Boston, MA, USA, 1988; pp. 1–14. ISBN 978-1-4684-5424-6. [Google Scholar]
- Hatfull, G.F. Bacteriophage Genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Lukianova, A.; Sykilinda, N.; Gorshkova, A.; Bondar, A.; Shneider, M.; Kabilov, M.; Drucker, V.; Miroshnikov, K. Pseudomonas Phage MD8: Genetic Mosaicism and Challenges of Taxonomic Classification of Lambdoid Bacteriophages. Int. J. Mol. Sci. 2021, 22, 10350. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, Y.; Zhang, J.; Yang, S.; Zhang, W. Multiple Novel Filamentous Phages Detected in the Cloacal Swab Samples of Birds Using Viral Metagenomics Approach. Virol. J. 2021, 18, 240. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.A.; Tan, C.H.; Mikkelsen, P.J.; Kung, V.; Woo, J.; Tay, M.; Hauser, A.; McDougald, D.; Webb, J.S.; Kjelleberg, S. The Biofilm Life Cycle and Virulence of Pseudomonas Aeruginosa Are Dependent on a Filamentous Prophage. ISME J. 2009, 3, 271–282. [Google Scholar] [CrossRef]
- Janmey, P.A.; Slochower, D.R.; Wang, Y.-H.; Wen, Q.; Cebers, A. Polyelectrolyte Properties of Filamentous Biopolymers and Their Consequences in Biological Fluids. Soft Matter 2014, 10, 1439–1449. [Google Scholar] [CrossRef]
- Winsor, G.L.; Griffiths, E.J.; Lo, R.; Dhillon, B.K.; Shay, J.A.; Brinkman, F.S.L. Enhanced Annotations and Features for Comparing Thousands of Pseudomonas Genomes in the Pseudomonas Genome Database. Nucleic Acids Res. 2016, 44, D646–D653. [Google Scholar] [CrossRef]
- Huber, K.E.; Waldor, M.K. Filamentous Phage Integration Requires the Host Recombinases XerC and XerD. Nature 2002, 417, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Levasseur, A.; La Scola, B.; Sharma, V.; Nasir, A.; Pontarotti, P.; Caetano-Anollés, G.; Raoult, D. Ancestrality and Mosaicism of Giant Viruses Supporting the Definition of the Fourth TRUC of Microbes. Front. Microbiol. 2018, 9, 2668. [Google Scholar] [CrossRef]
- Campbell, A. Comparative Molecular Biology of Lambdoid Phages. Annu. Rev. Microbiol. 1994, 48, 193–222. [Google Scholar] [CrossRef] [PubMed]
- Juhala, R.J.; Ford, M.E.; Duda, R.L.; Youlton, A.; Hatfull, G.F.; Hendrix, R.W. Genomic Sequences of Bacteriophages HK97 and HK022: Pervasive Genetic Mosaicism in the Lambdoid Bacteriophages. J. Mol. Biol. 2000, 299, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. MMBR 2004, 68, 560–602. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage Name | Accession | Phage ICTV Classification or Bacterial Strain for Prophages | Genome (Prophage Region) Length, Nucleotides | GC-Content |
|---|---|---|---|---|
| Pf1 | NC_001331.1 | Primolicivirus Pf1 | 7349 | 61.5% |
| Pf3 | M11912.1 | Tertilicivirus Pf3 | 5833 | 45.4% |
| Pf8 | MN710383.1 | Primolicivirus Pf8 | 10,061 | 58.1% |
| Pf4 prophage | AE004091.2 | P. aeruginosa PAO1 | 12,437 | 56.4% |
| Pf5 prophage | CP000438.1 | P. aeruginosa UCBPP-PA14 | 12,209 | 58.3% |
| Pf6 prophage | GQ141978.1 | P. aeruginosa PAO1 substrain MPAO1 | 12,066 | 55.9% |
| Pf7 prophage | CP000744.1 | P. aeruginosa PA7 | 12,933 | 56.8% |
| Pf-LESB58 prophage | FM209186.1 | P. aeruginosa LESB58 | 7599 | 60.5% |
| Pf Nhmuc prophage | CP013479.1 | P. aeruginosa NHmuc | 11,507 | 58.2% |
| Pf paerg010 | NZ_LR130536.1 | P. aeruginosa paerg010 | 10,682 | 56.3% |
| Pf paerg010-2 | NZ_LR130536.1 | P. aeruginosa paerg010 | 13,041 | 54.9% |
| Group 1 | Group 2 | Group 3 | Group 4 | Group 5 | Group 6 | Group 7 | |
|---|---|---|---|---|---|---|---|
| Number of found prophages regions | 34 | 28 | 2 | 3 | 4 | 1 | 1 |
| Isolate containing the representative prophage | 54-4 | 99-2 | 62-2 | 36-2 | 42-1 | 97-1 | 17,897 |
| Representative prophage region size, nucleotides | 13,193 | 13,538 | 12,602 | 7101 | 7418 | 6654 | 7182 |
| Representative prophage GC-content | 55.8% | 54.7% | 55.6% | 58.8% | 58.9% | 59.5% | 55.6% |
| Bacterial host genome GC-content | 66.1% | 66.4% | 66.5% | 66.5% | 66.4% | 66.4% | 66.5% |
| Number of predicted ORFs in representative prophage | 18 | 18 | 15 | 10 | 9 | 10 | 12 |
| Size of terminal direct repeats in representative prophage, nucleotides | 66 | 82 | 51 | 12 | 12 | 11 | 12 |
| Presence of integrase in representative prophage | Yes | Yes | Yes | No | No | No | No |
| Integrations site in representative prophage | tRNA | tRNA | tRNA | intergenic region | intergenic region | intergenic region | intergenic region |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evseev, P.; Bocharova, J.; Shagin, D.; Chebotar, I. Analysis of Pseudomonas aeruginosa Isolates from Patients with Cystic Fibrosis Revealed Novel Groups of Filamentous Bacteriophages. Viruses 2023, 15, 2215. https://doi.org/10.3390/v15112215
Evseev P, Bocharova J, Shagin D, Chebotar I. Analysis of Pseudomonas aeruginosa Isolates from Patients with Cystic Fibrosis Revealed Novel Groups of Filamentous Bacteriophages. Viruses. 2023; 15(11):2215. https://doi.org/10.3390/v15112215
Chicago/Turabian StyleEvseev, Peter, Julia Bocharova, Dmitriy Shagin, and Igor Chebotar. 2023. "Analysis of Pseudomonas aeruginosa Isolates from Patients with Cystic Fibrosis Revealed Novel Groups of Filamentous Bacteriophages" Viruses 15, no. 11: 2215. https://doi.org/10.3390/v15112215