
A Re-Evaluation of African Swine Fever Genotypes Based on p72 Sequences Reveals the Existence of Only Six Distinct p72 Groups

 , , ,
, , ,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Description of the Dataset
2.2. Data Preprocessing
2.3. Data Analysis
3. Results and Discussion
3.1. P72 Sequences on NCBI
3.2. Examination of All the Full-Length p72 Sequences within the p72 ORF in the Database
3.3. Determining Grouping of p72 according to the Amino Acid Sequence
3.4. Analysis of Partial p72 Sequences Available on NCBI
3.5. ASFV Can Be Classified into Six Genotypes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Penrith, M.L.; Kivaria, F.M.; Masembe, C. One hundred years of African swine fever: A tribute to R. Eustace Montgomery. Transbound. Emerg. Dis. 2021, 68, 2640–2642. [Google Scholar] [CrossRef] [PubMed]
- Chapman, D.A.; Darby, A.C.; Da Silva, M.; Upton, C.; Radford, A.D.; Dixon, L.K. Genomic analysis of highly virulent Georgia 2007/1 isolate of African swine fever virus. Emerg. Infect. Dis. 2011, 17, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Ambagala, A.; Goonewardene, K.; Lamboo, L.; Goolia, M.; Erdelyan, C.; Fisher, M.; Handel, K.; Lung, O.; Blome, S.; King, J.; et al. Characterization of a Novel African Swine Fever Virus p72 Genotype II from Nigeria. Viruses 2023, 15, 915. [Google Scholar] [CrossRef] [PubMed]
- Spinard, E.; Rai, A.; Osei-Bonsu, J.; O’Donnell, V.; Ababio, P.T.; Tawiah-Yingar, D.; Arthur, D.; Baah, D.; Ramirez-Medina, E.; Espinoza, N.; et al. The 2022 Outbreaks of African Swine Fever Virus Demonstrate the First Report of Genotype II in Ghana. Viruses 2023, 15, 1722. [Google Scholar] [CrossRef] [PubMed]
- Okwasiimire, R.; Flint, J.F.; Kayaga, E.B.; Lakin, S.; Pierce, J.; Barrette, R.W.; Faburay, B.; Ndoboli, D.; Ekakoro, J.E.; Wampande, E.M.; et al. Whole Genome Sequencing Shows that African Swine Fever Virus Genotype IX Is Still Circulating in Domestic Pigs in All Regions of Uganda. Pathogens 2023, 12, 912. [Google Scholar] [CrossRef] [PubMed]
- Bisimwa, P.N.; Ongus, J.R.; Tiambo, C.K.; Machuka, E.M.; Bisimwa, E.B.; Steinaa, L.; Pelle, R. First detection of African swine fever (ASF) virus genotype X and serogroup 7 in symptomatic pigs in the Democratic Republic of Congo. Virol. J. 2020, 17, 135. [Google Scholar] [CrossRef]
- Adeola, A.C.; Luka, P.D.; Jiang, X.X.; Cai, Z.F.; Oluwole, O.O.; Shi, X.; Oladele, B.M.; Olorungbounmi, T.O.; Boladuro, B.; Omotosho, O.; et al. Target capture sequencing for the first Nigerian genotype I ASFV genome. Microb. Genom. 2023, 9, 001069. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrin, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a New Genotype of African Swine Fever Virus in Domestic Pigs from Ethiopia. Transbound. Emerg. Dis. 2017, 64, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Zani, L.; Forth, J.H.; Forth, L.; Nurmoja, I.; Leidenberger, S.; Henke, J.; Carlson, J.; Breidenstein, C.; Viltrop, A.; Hoper, D.; et al. Deletion at the 5’-end of Estonian ASFV strains associated with an attenuated phenotype. Sci. Rep. 2018, 8, 6510. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Camos, L.; Alonso, U.; Esteve-Codina, A.; Chang, C.Y.; Martin-Mur, B.; Accensi, F.; Munoz, M.; Navas, M.J.; Dabad, M.; Vidal, E.; et al. Cross-protection against African swine fever virus upon intranasal vaccination is associated with an adaptive-innate immune crosstalk. PLoS Pathog. 2022, 18, e1010931. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, P.L.; Lacasta, A.; Lopez, E.; Bosch, L.; Collado, J.; Pina-Pedrero, S.; Correa-Fiz, F.; Accensi, F.; Navas, M.J.; Vidal, E.; et al. BA71DeltaCD2: A New Recombinant Live Attenuated African Swine Fever Virus with Cross-Protective Capabilities. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.D.; Penrith, M.L.; Cruciere, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Lubisi, B.A.; Bastos, A.D.; Dwarka, R.M.; Vosloo, W. Molecular epidemiology of African swine fever in East Africa. Arch. Virol. 2005, 150, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- Zsak, L.; Borca, M.V.; Risatti, G.R.; Zsak, A.; French, R.A.; Lu, Z.; Kutish, G.F.; Neilan, J.G.; Callahan, J.D.; Nelson, W.M.; et al. Preclinical diagnosis of African swine fever in contact-exposed swine by a real-time PCR assay. J. Clin. Microbiol. 2005, 43, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nunez, D.; Castillo-Rosa, E.; Vigara-Astillero, G.; Garcia-Belmonte, R.; Gallardo, C.; Revilla, Y. Identification and Isolation of Two Different Subpopulations within African Swine Fever Virus Arm/07 Stock. Vaccines 2020, 8, 625. [Google Scholar] [CrossRef] [PubMed]
- Masembe, C.; Phan, M.V.T.; Robertson, D.L.; Cotten, M. Increased resolution of African swine fever virus genome patterns based on profile HMMs of protein domains. Virus Evol. 2020, 6, veaa044. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
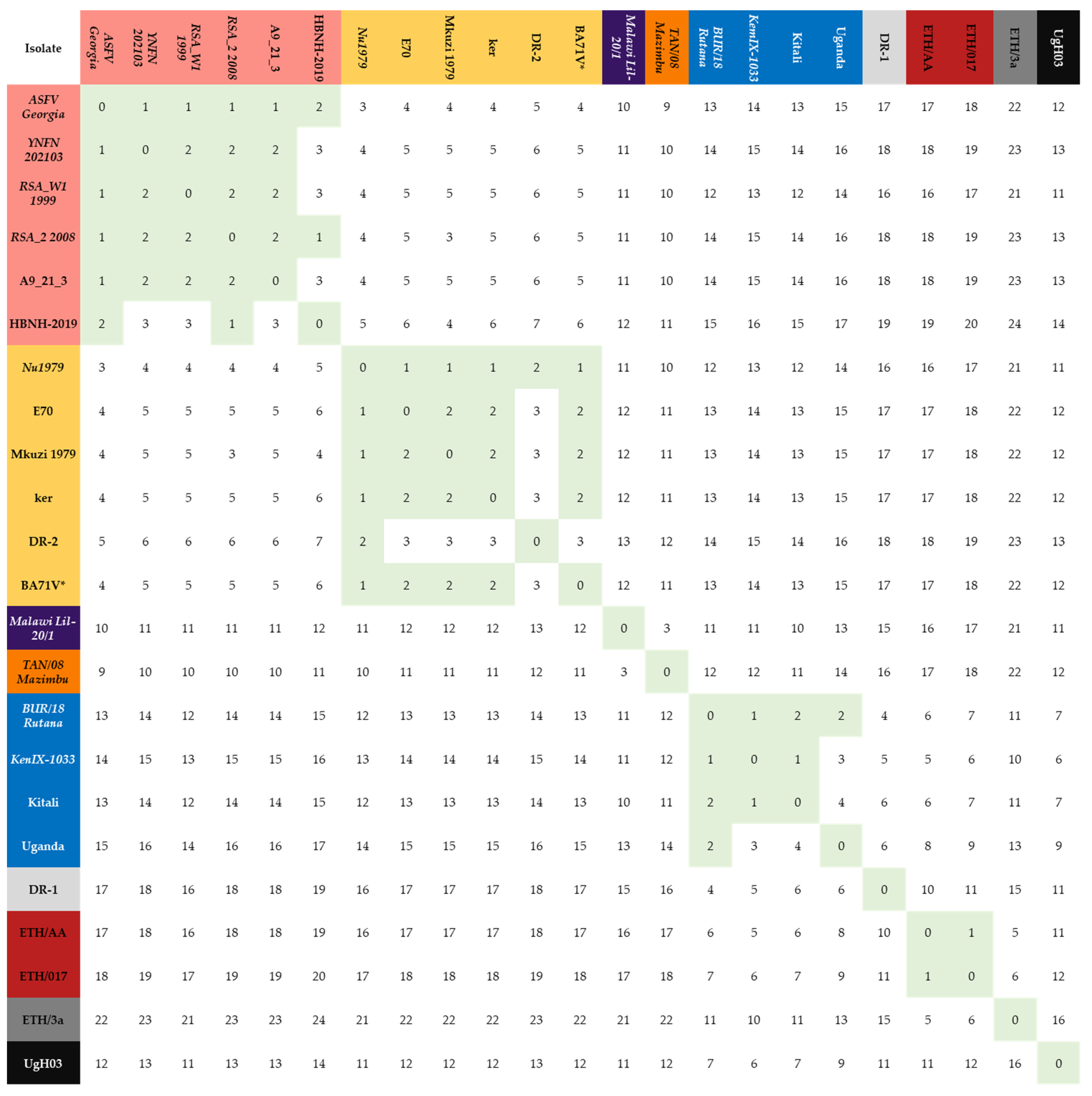
| Genotype 2 | Genotype 1 | Genotype 8 | Genotype 9 | Genotype 15 | Genotype 23 | |
|---|---|---|---|---|---|---|
| Genotype 2 | 1.9 | 4.9 | 11.0 | 14.4 | 10.0 | 18.2 |
| Genotype 1 | 4.9 | 2.0 | 12.0 | 13.8 | 11.0 | 17.5 |
| Genotype 8 | 11.0 | 12.0 | 0 | 11.3 | 3.0 | 16.5 |
| Genotype 9 | 14.4 | 13.8 | 11.3 | 2.17 | 12.3 | 6.8 |
| Genotype 15 | 10.0 | 11.0 | 3 | 12.3 | 0.0 | 17.5 |
| Genotype 23 | 18.2 | 17.5 | 16.5 | 6.8 | 17.5 | 1.0 |
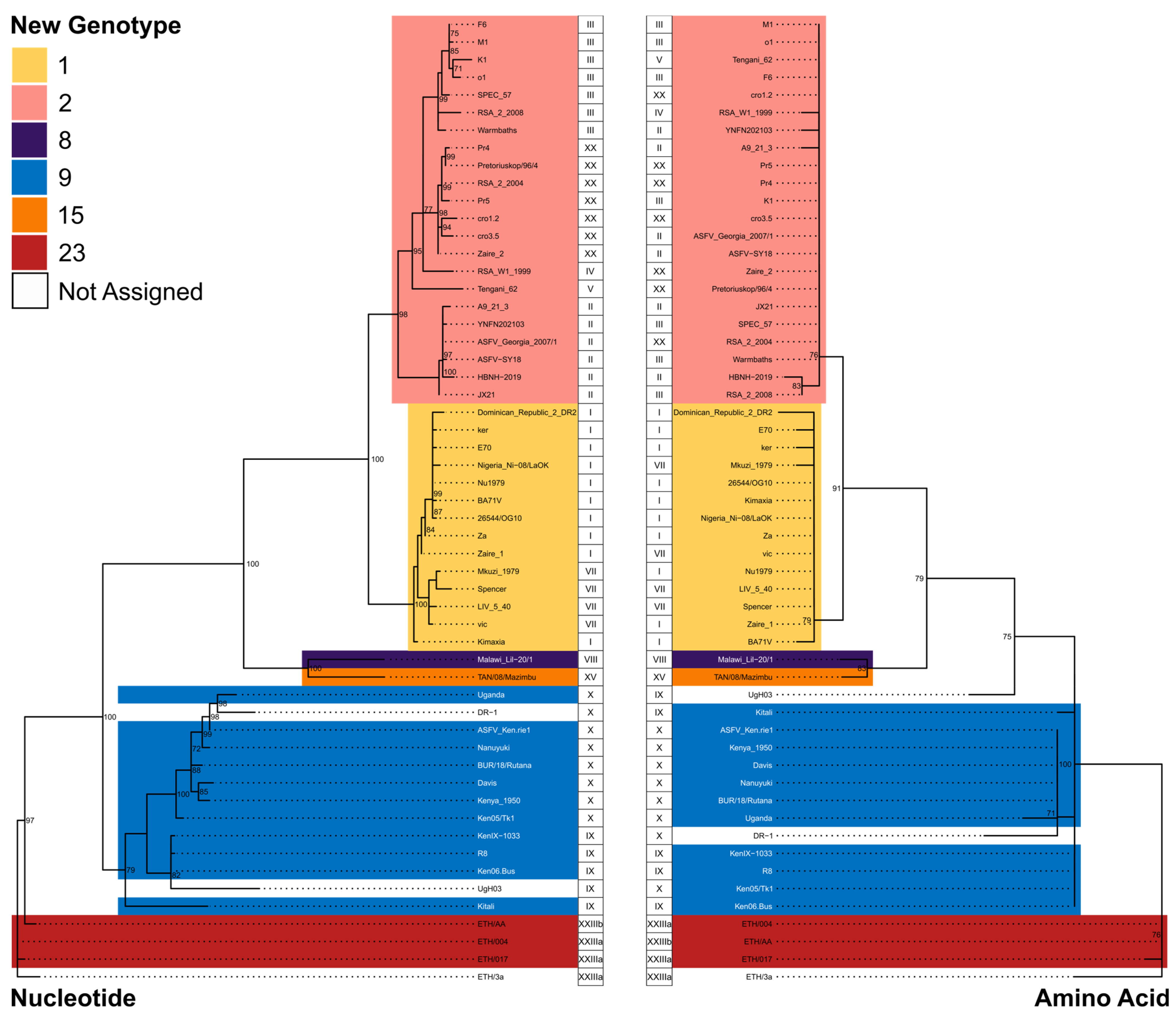
| New Genotype Assignment | Historical Genotype |
|---|---|
| 1 | I, VII, and XXII |
| 2 | II, III, IV, V, XVII, XX, XXI, and XXIV |
| 8 | VIII |
| 9 | IX and X |
| 15 | XV |
| 23 | XXIIIa and XXIIIb |
| New Genotype | Number of Isolates | Number of Unique Sequences |
|---|---|---|
| 1 | 142 | 18 |
| 2 | 224 | 34 |
| 8 | 5 | 3 |
| 9 | 297 | 30 |
| 15 | 4 | 2 |
| 23 | 10 | 5 |
| 1 or 2 | 1840 | 116 |
| 1 or 2 or 8 | 1 | 1 |
| 1 or 2 or 8 or 9 or 15 | 1 | 1 |
| 1 or 2 or 8 or 9 or 23 | 1 | 1 |
| 1 or 2 or 9 or 23 | 4 | 1 |
| 1 or 8 or 9 or 2 or 15 | 1 | 1 |
| 1 or 9 or 23 | 2 | 1 |
| 2 or 9 | 1 | 1 |
| 8 or 15 | 72 | 12 |
| 8 or 9 or 15 | 1 | 1 |
| 9 or 23 | 31 | 4 |
| not defined | 15 | 14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spinard, E.; Dinhobl, M.; Tesler, N.; Birtley, H.; Signore, A.V.; Ambagala, A.; Masembe, C.; Borca, M.V.; Gladue, D.P. A Re-Evaluation of African Swine Fever Genotypes Based on p72 Sequences Reveals the Existence of Only Six Distinct p72 Groups. Viruses 2023, 15, 2246. https://doi.org/10.3390/v15112246
Spinard E, Dinhobl M, Tesler N, Birtley H, Signore AV, Ambagala A, Masembe C, Borca MV, Gladue DP. A Re-Evaluation of African Swine Fever Genotypes Based on p72 Sequences Reveals the Existence of Only Six Distinct p72 Groups. Viruses. 2023; 15(11):2246. https://doi.org/10.3390/v15112246
Chicago/Turabian StyleSpinard, Edward, Mark Dinhobl, Nicolas Tesler, Hillary Birtley, Anthony V. Signore, Aruna Ambagala, Charles Masembe, Manuel V. Borca, and Douglas P. Gladue. 2023. "A Re-Evaluation of African Swine Fever Genotypes Based on p72 Sequences Reveals the Existence of Only Six Distinct p72 Groups" Viruses 15, no. 11: 2246. https://doi.org/10.3390/v15112246
APA StyleSpinard, E., Dinhobl, M., Tesler, N., Birtley, H., Signore, A. V., Ambagala, A., Masembe, C., Borca, M. V., & Gladue, D. P. (2023). A Re-Evaluation of African Swine Fever Genotypes Based on p72 Sequences Reveals the Existence of Only Six Distinct p72 Groups. Viruses, 15(11), 2246. https://doi.org/10.3390/v15112246