
A Novel Subcluster of Closely Related Bacillus Phages with Distinct Tail Fiber/Lysin Gene Combinations
 , , , ,
, , , ,
Abstract
:1. Introduction
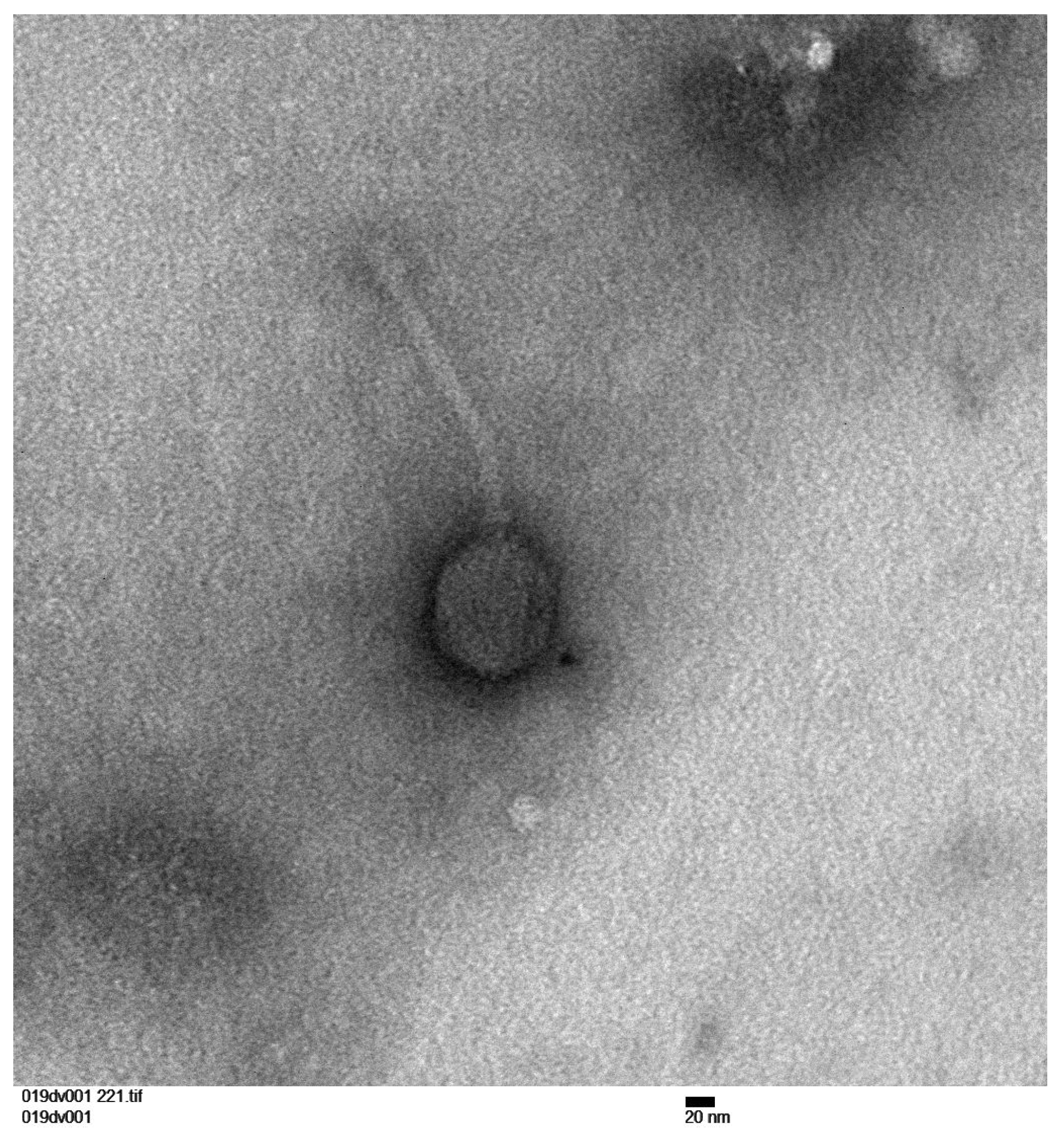
2. Materials and Methods
3. Results
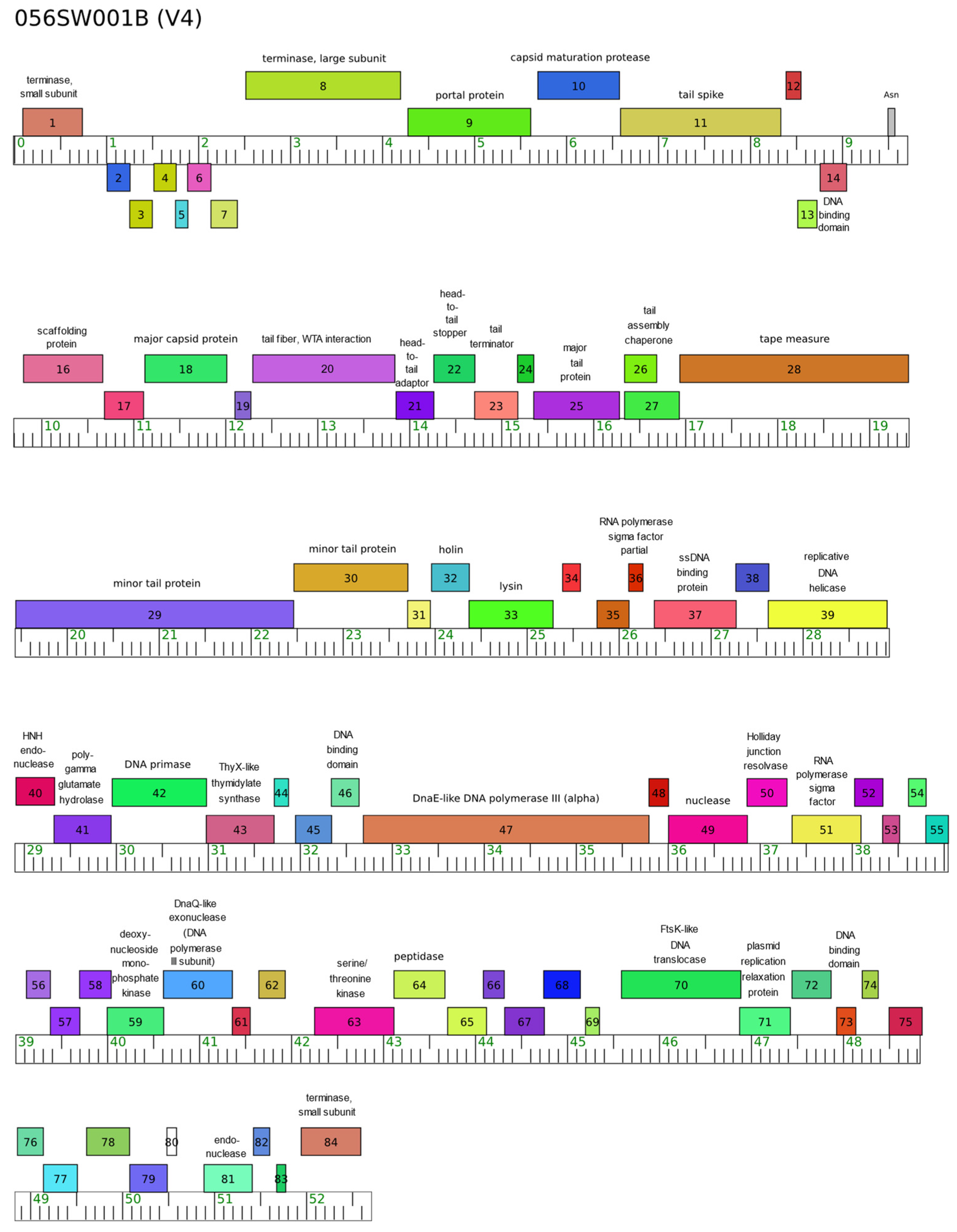
3.1. Basic Genomic Data
3.2. Horizontal Gene Transfer in Subcluster V4 Evolution
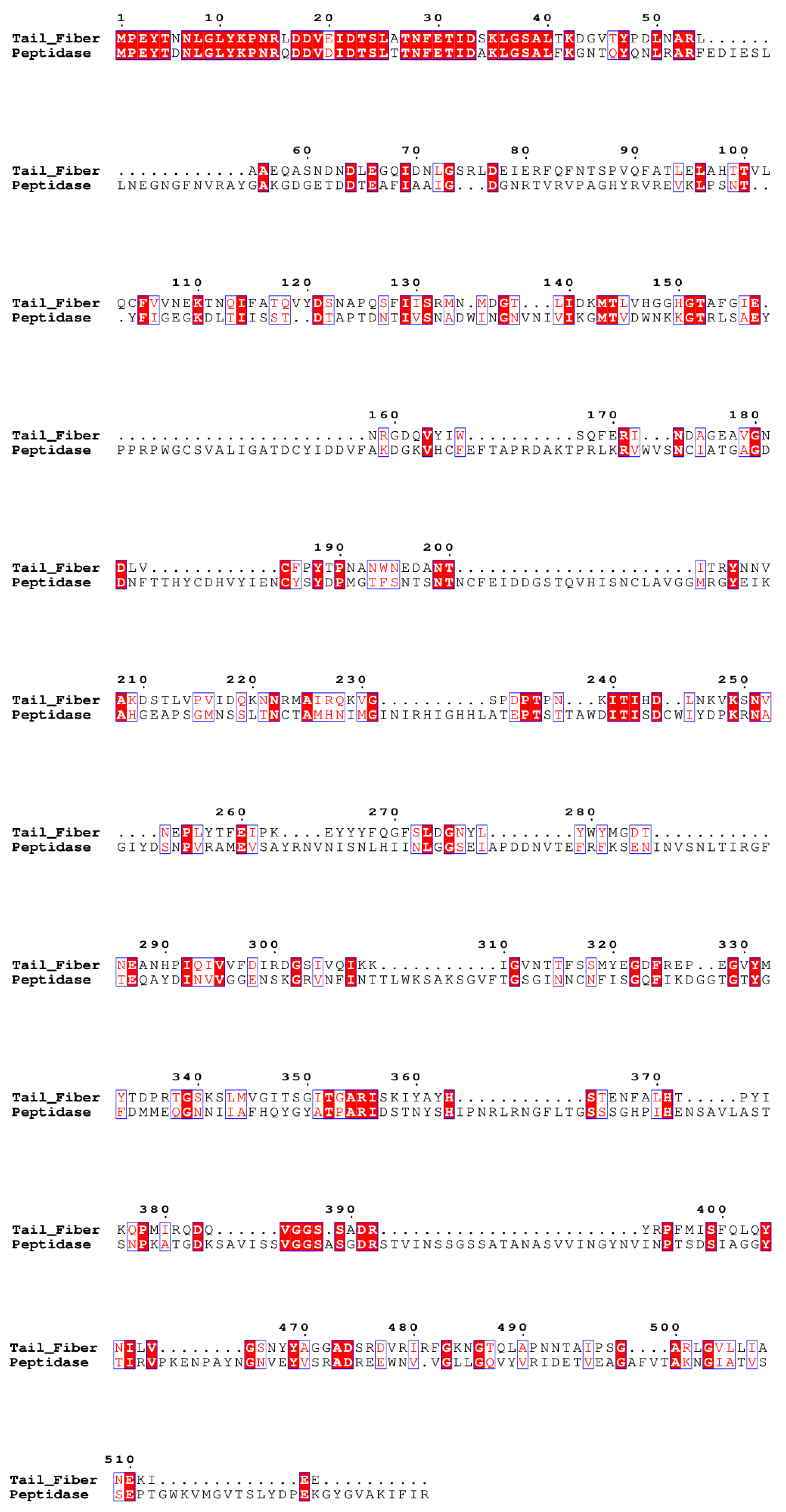
3.2.1. Tail Fiber
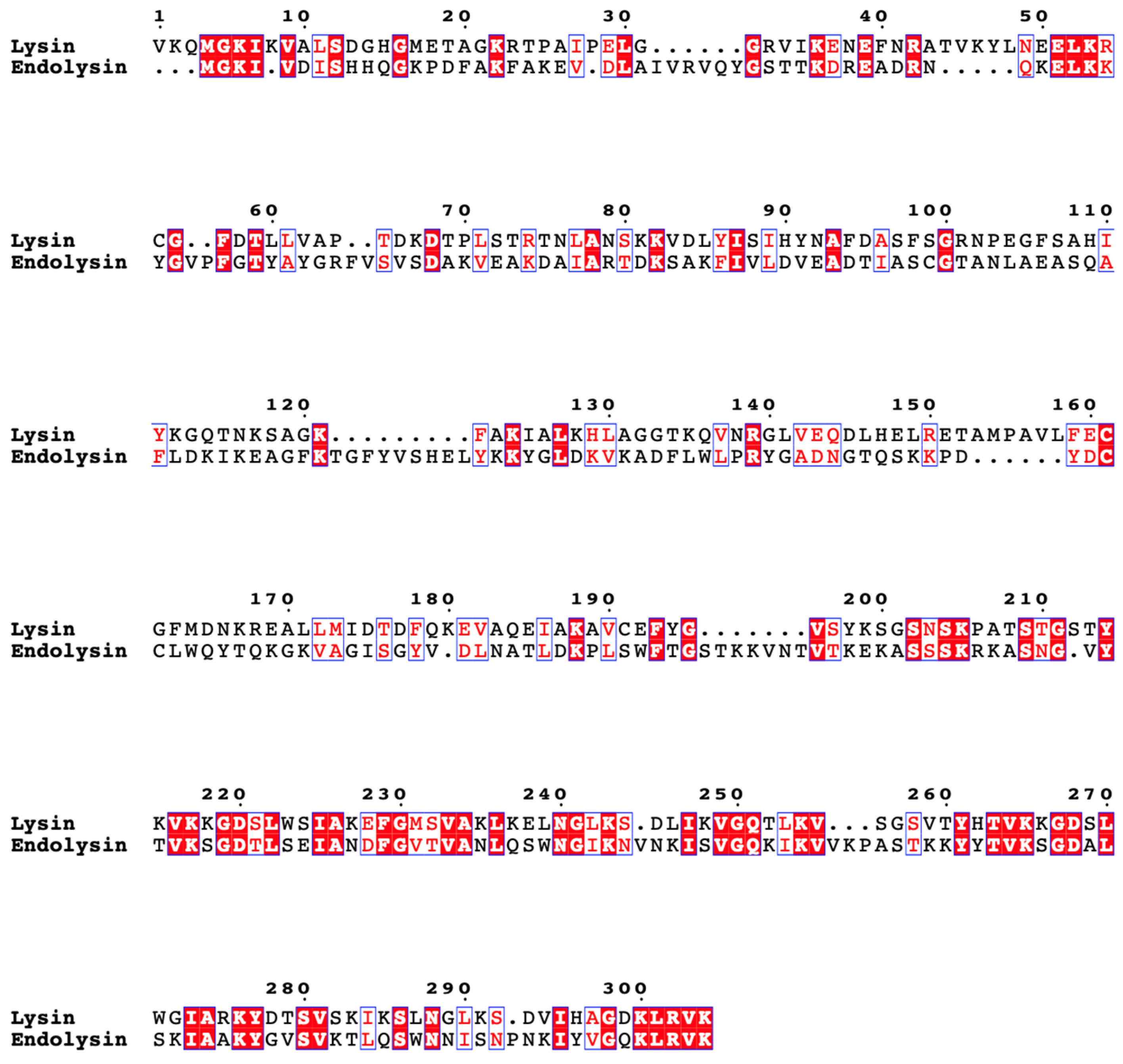
3.2.2. Lysin
3.2.3. Other Genes
3.3. Host Range
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koskella, B.; Brockhurst, M.A. Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Dy, R.L.; Richter, C.; Salmond, G.P.C.; Fineran, P.C. Remarkable Mechanisms in Microbes to Resist Phage Infections. Annu. Rev. Virol. 2014, 1, 307–331. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.; Brockhurst, M.A. Ecological and Evolutionary Benefits of Temperate Phage: What Does or Doesn’t Kill You Makes You Stronger. BioEssays 2017, 39, 1700112. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Hernandez, C.A.; Wheatley, R.M. Understanding the Impacts of Bacteriophage Viruses: From Laboratory Evolution to Natural Ecosystems. Annu. Rev. Virol. 2022, 9, 57–78. [Google Scholar] [CrossRef]
- Brown, T.L.; Charity, O.J.; Adriaenssens, E.M. Ecological and functional roles of bacteriophages in contrasting environments: Marine, terrestrial and human gut. Curr. Opin. Microbiol. 2022, 70, 102229. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Puxty, R.J.; Millard, A.D.; Evans, D.J.; Scanlan, D.J. Viruses Inhibit CO2 Fixation in the Most Abundant Phototrophs on Earth. Curr. Biol. 2016, 26, 1585–1589. [Google Scholar] [CrossRef]
- Howard-Varona, C.; Lindback, M.M.; Bastien, G.E.; Solonenko, N.; Zayed, A.A.; Jang, H.; Andreopoulos, B.; Brewer, H.M.; Glavina Del Rio, T.; Adkins, J.N.; et al. Phage-specific metabolic reprogramming of virocells. ISME J. 2020, 14, 881–895. [Google Scholar] [CrossRef]
- Borodovich, T.; Shkoporov, A.N.; Ross, R.P.; Hill, C. Phage-mediated horizontal gene transfer and its implications for the human gut microbiome. Gastroenterol. Rep. 2022, 10, goac012. [Google Scholar] [CrossRef]
- Ma, Y.; You, X.; Mai, G.; Tokuyasu, T.; Liu, C. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome 2018, 6, 24. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Dark Matter of the Biosphere: The Amazing World of Bacteriophage Diversity. J. Virol. 2015, 89, 8107–8110. [Google Scholar] [CrossRef] [PubMed]
- Bibby, K. Improved Bacteriophage Genome Data is Necessary for Integrating Viral and Bacterial Ecology. Microb. Ecol. 2014, 67, 242–244. [Google Scholar] [CrossRef]
- Comeau, A.M.; Hatfull, G.F.; Krisch, H.M.; Lindell, D.; Mann, N.H.; Prangishvili, D. Exploring the prokaryotic virosphere. Res. Microbiol. 2008, 159, 306–313. [Google Scholar] [CrossRef]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Jacobs-Sera, D.; Lawrence, J.G.; Pope, W.H.; Russell, D.A.; Ko, C.-C.; Weber, R.J.; Patel, M.C.; Germane, K.L.; Edgar, R.H.; et al. Comparative Genomic Analysis of 60 Mycobacteriophage Genomes: Genome Clustering, Gene Acquisition, and Gene Size. J. Mol. Biol. 2010, 397, 119–143. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Pedulla, M.L.; Jacobs-Sera, D.; Cichon, P.M.; Foley, A.; Ford, M.E.; Gonda, R.M.; Houtz, J.M.; Hryckowian, A.J.; Kelchner, V.A.; et al. Exploring the Mycobacteriophage Metaproteome: Phage Genomics as an Educational Platform. PLoS Genet. 2006, 2, e92. [Google Scholar] [CrossRef]
- Payne, K.M.; Hatfull, G.F. Mycobacteriophage Endolysins: Diverse and Modular Enzymes with Multiple Catalytic Activities. PLoS ONE 2012, 7, e34052. [Google Scholar] [CrossRef]
- Delesalle, V.A.; Tomko, B.E.; Vill, A.C.; Lichty, K.B.; Krukonis, G.P. Forty Years without Family: Three Novel Bacteriophages with High Similarity to SPP1 Reveal Decades of Evolutionary Stasis since the Isolation of Their Famous Relative. Viruses 2022, 14, 2106. [Google Scholar] [CrossRef]
- Vill, A.C.; Delesalle, V.A.; Tomko, B.E.; Lichty, K.B.; Strine, M.S.; Guffey, A.A.; Burton, E.A.; Tanke, N.T.; Krukonis, G.P. Comparative Genomics of Six Lytic Bacillus subtilis Phages from the Southwest United States. PHAGE 2022, 3, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Krukonis, G.P.; Kemp, A.K.; Storrie, K.F.; Chavira, V.R.; Lantrip, H.W.; Perez, V.D.; Reyes, D.A.; Truax, J.A.; Loney, R.; Delesalle, V.A. Complete Genome Sequences of Two Temperate Bacillus subtilis Phages Isolated at Tumamoc Hill Desert Laboratory. Microbiol. Resour. Announc. 2022, 11, e00455-22. [Google Scholar] [CrossRef] [PubMed]
- Earl, A.M.; Losick, R.; Kolter, R. Ecology and genomics of Bacillus subtilis. Trends Microbiol. 2008, 16, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Chanal, A.; Chapon, V.; Benzerara, K.; Barakat, M.; Christen, R.; Achouak, W.; Barras, F.; Heulin, T. The desert of Tataouine: An extreme environment that hosts a wide diversity of microorganisms and radiotolerant bacteria. Environ. Microbiol. 2006, 8, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Prestel, E.; Salamitou, S.; DuBow, M.S. An examination of the bacteriophages and bacteria of the Namib desert. J. Microbiol. 2008, 46, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Prestel, E.; Regeard, C.; Salamitou, S.; Neveu, J.; DuBow, M.S. The bacteria and bacteriophages from a Mesquite Flats site of the Death Valley desert. Antonie Van Leeuwenhoek 2013, 103, 1329–1341. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Hashem, A.; Abd_Allah, E.F. Bacillus: A Biological Tool for Crop Improvement through Bio-Molecular Changes in Adverse Environments. Front. Physiol. 2017, 8, 667. [Google Scholar] [CrossRef]
- Viju, N.; Punitha, S.M.J.; Satheesh, S. Antibiofilm activity of symbiotic Bacillus species associated with marine gastropods. Ann. Microbiol. 2020, 70, 11. [Google Scholar] [CrossRef]
- Melara, E.G.; Avellaneda, M.C.; Valdivié, M.; García-Hernández, Y.; Aroche, R.; Martínez, Y. Probiotics: Symbiotic Relationship with the Animal Host. Animals 2022, 12, 719. [Google Scholar] [CrossRef]
- Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Imbroglios of Viral Taxonomy: Genetic Exchange and Failings of Phenetic Approaches. J. Bacteriol. 2002, 184, 4891–4905. [Google Scholar] [CrossRef]
- Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Karthikeyan, T.; Wadsworth, C.; Lewis, J.A.; Jacobs-Sera, D.; Falbo, J.; Gross, J.; Pannunzio, N.R.; et al. Origins of Highly Mosaic Mycobacteriophage Genomes. Cell 2003, 113, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Brussow, H.; Desiere, F. Comparative phage genomics and the evolution of Siphoviridae: Insights from dairy phages. Mol. Microbiol. 2001, 39, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Belcaid, M.; Bergeron, A.; Poisson, G. Mosaic Graphs and Comparative Genomics in Phage Communities. J. Comput. Biol. 2010, 17, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2017, 2, 17112. [Google Scholar] [CrossRef]
- Duncan, K.E.; Ferguson, N.; Kimura, K.; Zhou, X.; Istock, C.A. Fine-scale genetic and phenotypic structure in natural populations of Bacillus subtilis and Bacillus licheniformis: Implications for bacterial evolution and speciation. Evolution 1994, 48, 2002–2025. [Google Scholar] [CrossRef]
- Istock, C.A.; Ferguson, N.; Istock, N.L.; Duncan, K.E. Geographical diversity of genomic lineages in Bacillus subtilis (Ehrenberg) Cohn sensu lato. Org. Divers. Evol. 2001, 1, 179–191. [Google Scholar] [CrossRef]
- The Actinobacteriophage Database|Protocols for Extraction. Available online: https://phagesdb.org/workflow/Extraction/ (accessed on 19 August 2023).
- Russell, D.A. Sequencing, Assembling, and Finishing Complete Bacteriophage Genomes. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Lavigne, R., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1681, pp. 109–125. ISBN 978-1-4939-7341-5. [Google Scholar] [CrossRef]
- Gordon, D.; Green, P. Consed: A graphical editor for next-generation sequencing. Bioinformatics 2013, 29, 2936–2937. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Lawrence, J.G. DNAMaster. Available online: http://cobamide2.bio.pitt.edu/computer.htm (accessed on 16 September 2023).
- Delcher, A. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef]
- Lukashin, A. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef]
- Altschu, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed]
- Cresawn, S.G.; Bogel, M.; Day, N.; Jacobs-Sera, D.; Hendrix, R.W.; Hatfull, G.F. Phamerator: A bioinformatic tool for comparative bacteriophage genomics. BMC Bioinform. 2011, 12, 395. [Google Scholar] [CrossRef]
- Gauthier, C.H.; Cresawn, S.G.; Hatfull, G.F. PhaMMseqs: A new pipeline for constructing phage gene phamilies using MMseqs2. G3 GenesGenomesGenetics 2022, 12, jkac233. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The Enveomics Collection: A Toolbox for Specialized Analyses of Microbial Genomes and Metagenomes. PeerJ Prepr 2016. Available online: https://peerj.com/preprints/1900v1 (accessed on 16 September 2023).
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Xie, Y.; Wahab, L.; Gill, J. Development and Validation of a Microtiter Plate-Based Assay for Determination of Bacteriophage Host Range and Virulence. Viruses 2018, 10, 189. [Google Scholar] [CrossRef]
- Bacillus Genetic Stock Center. Available online: https://bgsc.org/ (accessed on 15 September 2023).
- Kunst, F.; Ogasawara, N.; Moszer, I.; Albertini, A.M.; Alloni, G.; Azevedo, V.; Bertero, M.G.; Bessières, P.; Bolotin, A.; Borchert, S.; et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 1997, 390, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Koç, C.; Xia, G.; Kühner, P.; Spinelli, S.; Roussel, A.; Cambillau, C.; Stehle, T. Structure of the host-recognition device of Staphylococcus aureus phage ϕ11. Sci. Rep. 2016, 6, 27581. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, F.C.; Baddiley, J. A Continuum of Anionic Charge: Structures and Functions of d-Alanyl-Teichoic Acids in Gram-Positive Bacteria. Microbiol. Mol. Biol. Rev. 2003, 67, 686–723. [Google Scholar] [CrossRef]
- Brown, S.; Santa Maria, J.P.; Walker, S. Wall Teichoic Acids of Gram-Positive Bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef]
- Xia, G.; Corrigan, R.M.; Winstel, V.; Goerke, C.; Gründling, A.; Peschel, A. Wall Teichoic Acid-Dependent Adsorption of Staphylococcal Siphovirus and Myovirus. J. Bacteriol. 2011, 193, 4006–4009. [Google Scholar] [CrossRef] [PubMed]
- Baptista, C.; Santos, M.A.; São-José, C. Phage SPP1 Reversible Adsorption to Bacillus subtilis Cell Wall Teichoic Acids Accelerates Virus Recognition of Membrane Receptor YueB. J. Bacteriol. 2008, 190, 4989–4996. [Google Scholar] [CrossRef]
- Schulz, E.C.; Ficner, R. Knitting and snipping: Chaperones in β-helix folding. Curr. Opin. Struct. Biol. 2011, 21, 232–239. [Google Scholar] [CrossRef]
- Fischetti, V.A. Bacteriophage lysins as effective antibacterials. Curr. Opin. Microbiol. 2008, 11, 393–400. [Google Scholar] [CrossRef]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef]
- Edgell, D.R.; Gibb, E.A.; Belfort, M. Mobile DNA elements in T4 and related phages. Virol. J. 2010, 7, 290. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Bacteriophage Host Range and Bacterial Resistance. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 70, pp. 217–248. ISBN 978-0-12-380991-9. [Google Scholar] [CrossRef]
- Choudoir, M.J.; DeAngelis, K.M. A framework for integrating microbial dispersal modes into soil ecosystem ecology. iScience 2022, 25, 103887. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Blackman, S.A.; Foster, S.J. Autolysins of Bacillus subtilis: Multiple enzymes with multiple functions. Microbiology 2000, 146, 249–262. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Isolation Strain | Genome Size (bp) | % GC | No. of ORFs | Collecting Site and Date | GPS Coordinates | Accession |
|---|---|---|---|---|---|---|---|
| 019DV002 | T89-06 | 52,749 | 42.6 | 84 * | Death Valley NP, Ca, May 2014 | 36°27′51.4″ N, 117°14′08.6″ W | MN176220 |
| 019DV004 | T89-06 | 52,749 | 42.6 | 84 * | MN176221 | ||
| 056SW001B | T89-20 | 52,692 | 42.6 | 83 * | Saguaro West NP, AZ, May 2014 | 32°16′12.7″ N, 111°12′20.6″ W | MN176230 |
| 268TH004 | TT123 | 52,834 | 42.4 | 80 * | Tumamoc Hill, Tucson, AZ May 2016 | 32°13′04.9″ N, 111°00′12.9″ W | MW394467.1 |
| 274BB002 | TT123 | 52,372 | 42.5 | 81 * | Big Bend NP, TX, June 2016 | 29°15′54.6″ N, 103°07′24.5″ W | MZ501264 |
| 276BB001 | TT123 | 52,370 | 42.5 | 81 * | 29°18′16.6″ N, 103°15′52.9″ W | MN176231 | |
| 280BB001 | 168 | 52,371 | 42.5 | 81 * | 29°18′29.0″ N, 103°28′42.6″ W | MN176232 |
| 019DV002 | 019DV004 | 056SW001B | 268TH004 | 274BB002 | 276BB001 | 280BB001 | |
| 019DV002 | --- | 100 | 96.7 | 95.4 | 96.2 | 96.2 | 96.2 |
| 019DV004 | 100 | --- | 96.7 | 95.4 | 96.2 | 96.2 | 96.2 |
| 056SW001B | 94.6 | 94.8 | --- | 95.4 | 96.4 | 96.4 | 96.4 |
| 268TH004 | 91.2 | 92.3 | 93.2 | --- | 98.1 | 98.1 | 98.1 |
| 274BB002 | 92.3 | 92.7 | 92.4 | 95.5 | --- | 100 | 99.9 |
| 276BB001 | 92.4 | 92.8 | 92.4 | 95.3 | 100 | 100 | 100 |
| 280BB001 | 92.4 | 92.8 | 92.4 | 94.9 | 100 | 100 | --- |
| Bacterial Strain | |||||||
|---|---|---|---|---|---|---|---|
| Domesticated | Wild | ||||||
| Phage | 168 | W23 | TG115 | T89-05 | T89-06 | T89-20 | T89-30 |
| 019DV002 | Y* | Y | N | Y | Y | Y | Y |
| 019DV004 | Y* | Y | N | Y | Y | Y | N** |
| 056SW001B | Y | Y | N | Y | Y | Y | Y |
| 268TH004 | N | Y | N | Y | Y | Y | N |
| 274BB002 | N | Y | N | Y | Y | Y | N |
| 276BB001 | Y | N** | N | Y | N** | Y | N |
| 280BB001 | Y | Y | N | Y | Y | Y | N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loney, R.E.; Delesalle, V.A.; Chaudry, B.E.; Czerpak, M.; Guffey, A.A.; Goubet-McCall, L.; McCarty, M.; Strine, M.S.; Tanke, N.T.; Vill, A.C.; et al. A Novel Subcluster of Closely Related Bacillus Phages with Distinct Tail Fiber/Lysin Gene Combinations. Viruses 2023, 15, 2267. https://doi.org/10.3390/v15112267
Loney RE, Delesalle VA, Chaudry BE, Czerpak M, Guffey AA, Goubet-McCall L, McCarty M, Strine MS, Tanke NT, Vill AC, et al. A Novel Subcluster of Closely Related Bacillus Phages with Distinct Tail Fiber/Lysin Gene Combinations. Viruses. 2023; 15(11):2267. https://doi.org/10.3390/v15112267
Chicago/Turabian StyleLoney, Rachel E., Véronique A. Delesalle, Brianne E. Chaudry, Megan Czerpak, Alexandra A. Guffey, Leo Goubet-McCall, Michael McCarty, Madison S. Strine, Natalie T. Tanke, Albert C. Vill, and et al. 2023. "A Novel Subcluster of Closely Related Bacillus Phages with Distinct Tail Fiber/Lysin Gene Combinations" Viruses 15, no. 11: 2267. https://doi.org/10.3390/v15112267