
Structure-Based High-Throughput Virtual Screening and Molecular Dynamics Simulation for the Discovery of Novel SARS-CoV-2 NSP3 Mac1 Domain Inhibitors




Abstract
:1. Introduction
2. Results
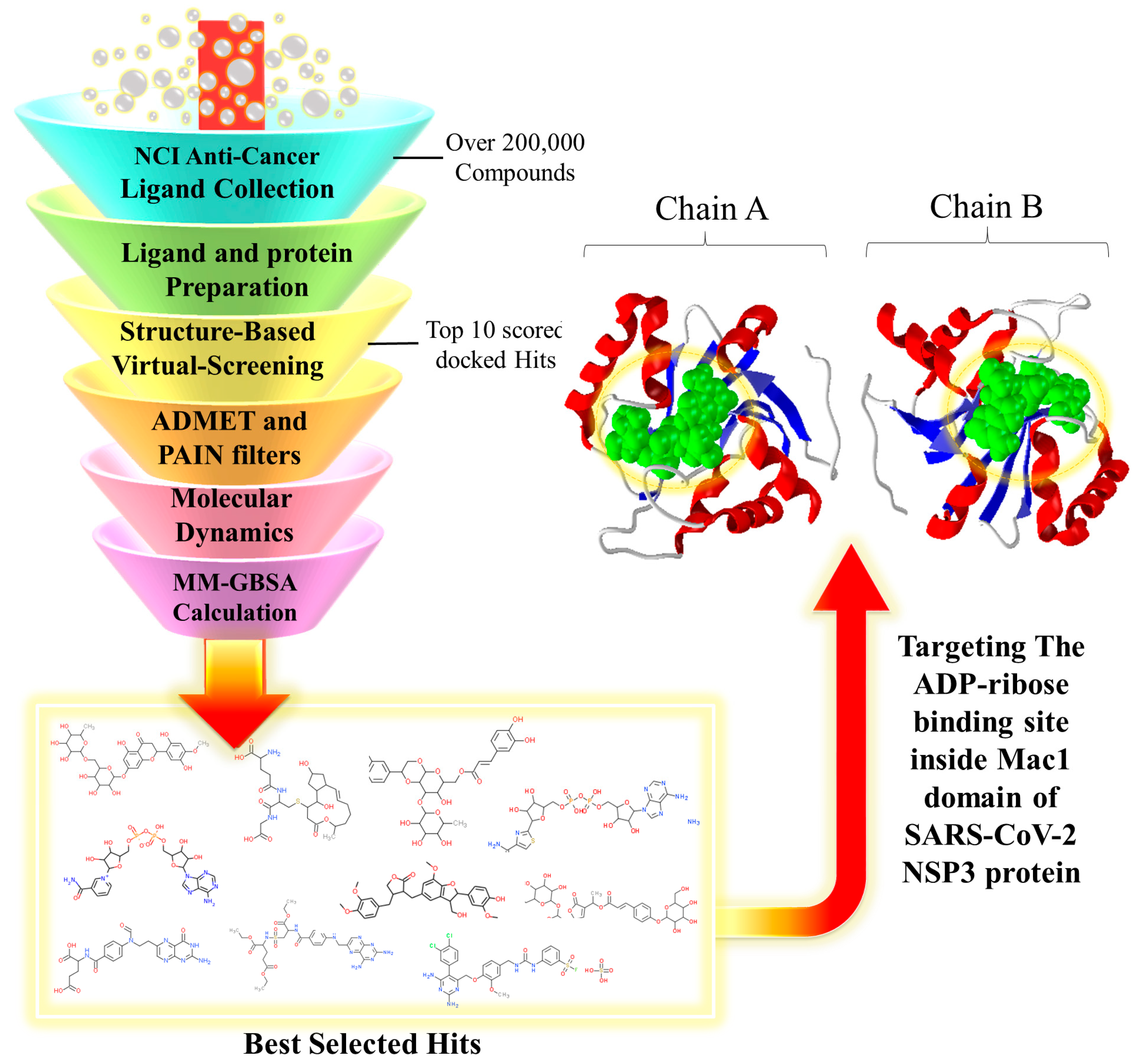
2.1. Structure-Based Virtual Screening
2.2. Physicochemical and Drug-like Properties
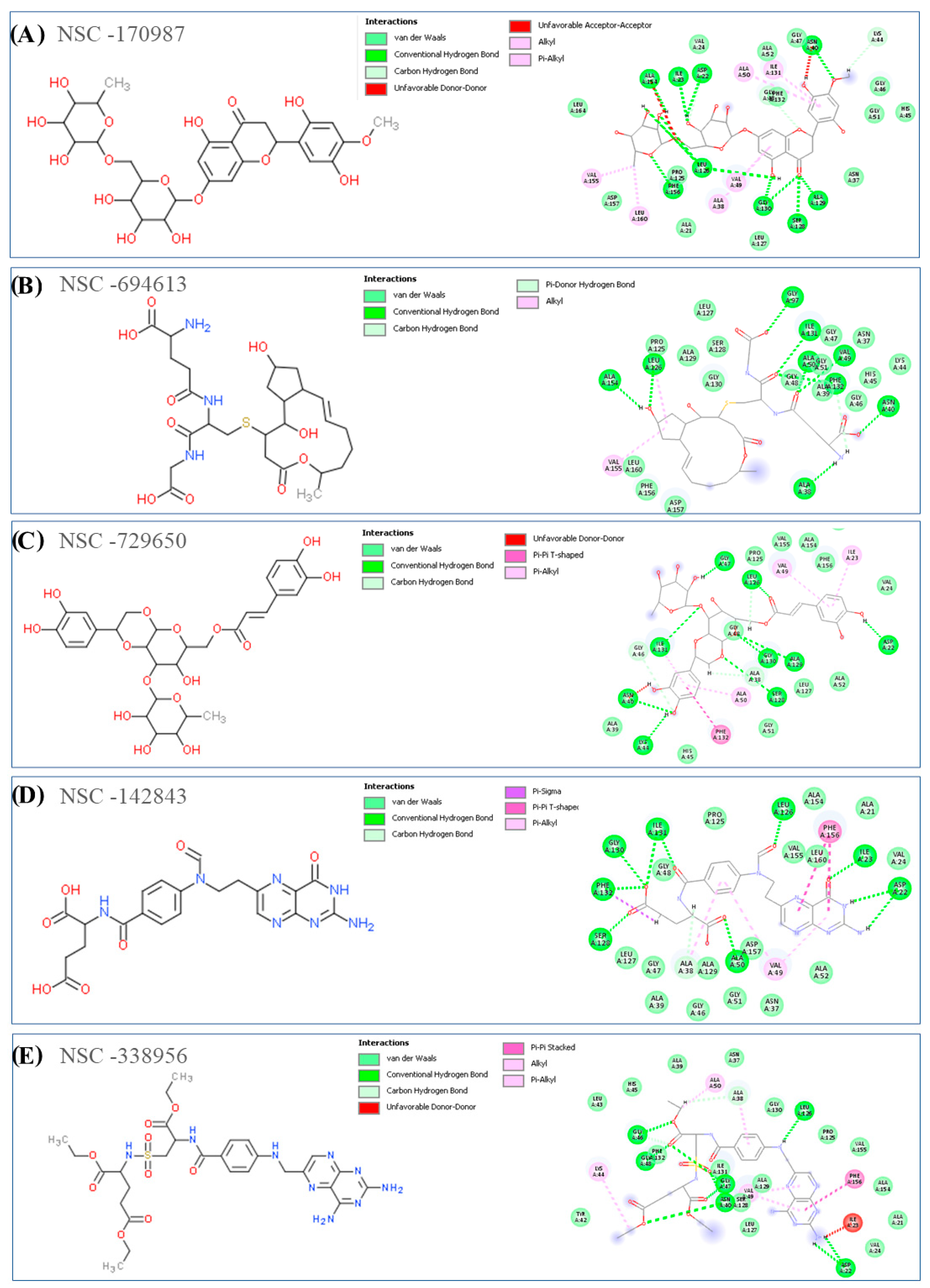
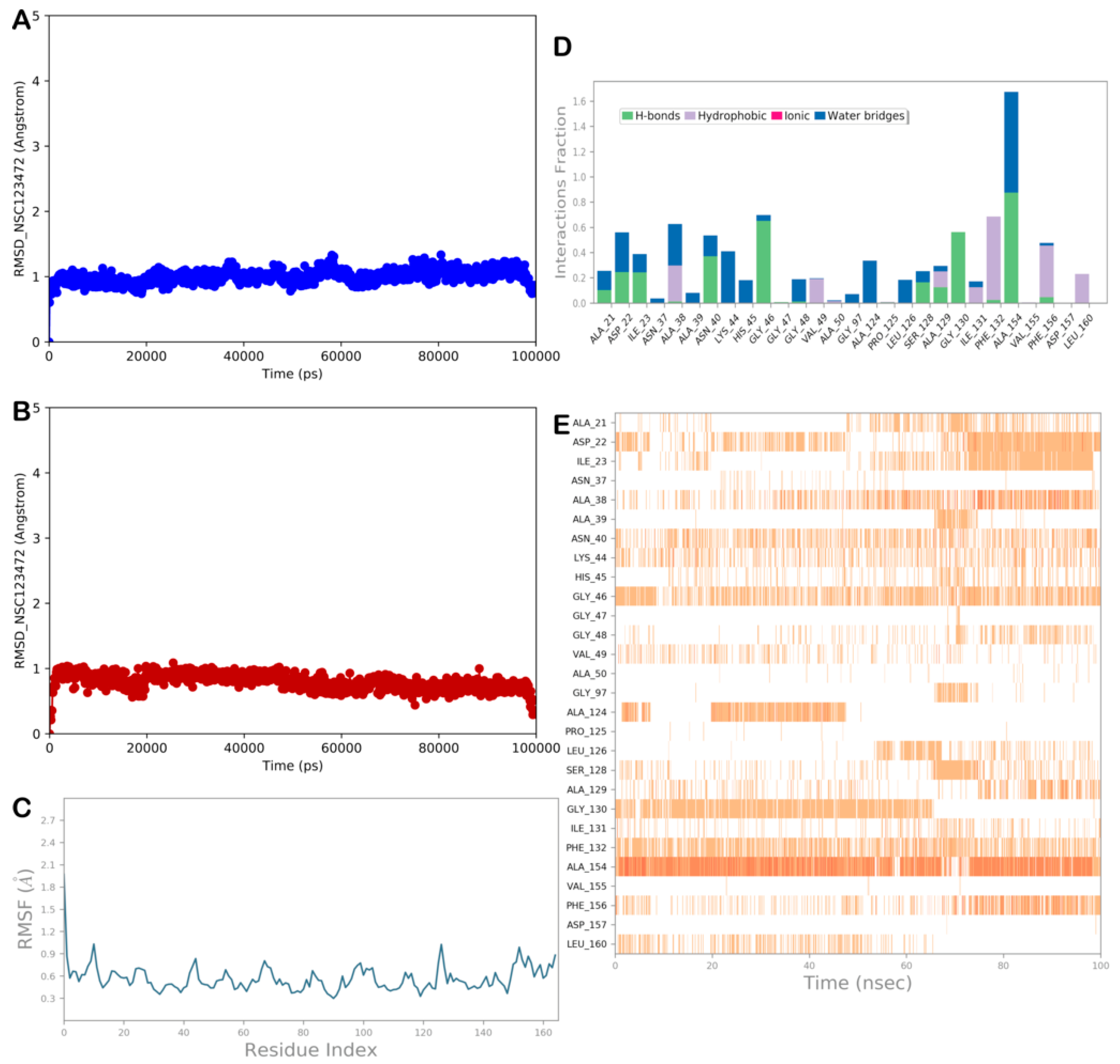
2.3. MD Simulation and MM/GBSA Analysis
2.3.1. MD Simulation Analysis of Compound NSC-358078
2.3.2. MD Simulation Analysis of Compound NSC-287067
2.3.3. MD Simulation Analysis of Compound NSC-142843
2.3.4. MD Simulation Analysis of Compound NSC-123472
3. Discussion
4. Materials and Methods
4.1. Selection of the Ligand Library
4.2. Structural Preparation of Protein and Ligands
4.3. Receptor Grid Generation
4.4. Structure-Based Virtual Screening
4.5. Analysis of ADMET and Physicochemical Properties
4.6. MD Simulation and MM/GBSA Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Gao, L. Chemistry and biology of SARS-CoV-2. Chem 2020, 6, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-Y.; Zhao, R.; Gao, L.-J.; Gao, X.-F.; Wang, D.-P.; Cao, J.-M. SARS-CoV-2: Structure, biology, and structure-based therapeutics development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- Telenti, A.; Hodcroft, E.B.; Robertson, D.L. The evolution and biology of SARS-CoV-2 variants. Cold Spring Harb. Perspect. Med. 2022, 12, a041390. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Zhong, Q.; Gao, G.F. Overview of SARS-CoV-2 genome-encoded proteins. Sci. China Life Sci. 2022, 65, 280–294. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921.e910. [Google Scholar] [CrossRef]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal Transduct. Target. Ther. 2022, 7, 1–28. [Google Scholar] [CrossRef]
- Rohaim, M.A.; El Naggar, R.F.; Clayton, E.; Munir, M. Structural and functional insights into non-structural proteins of coronaviruses. Microb. Pathog. 2021, 150, 104641. [Google Scholar] [CrossRef]
- Rezaei, S.; Sefidbakht, Y. Structure of SARS-CoV-2 Proteins. In COVID-19; Rahmandoust, M., Ranaei-Siadat, S.O., Eds.; Springer: Singapore, 2021; pp. 91–120. [Google Scholar] [CrossRef]
- Yan, F.; Gao, F. An overview of potential inhibitors targeting non-structural proteins 3 (PLpro and Mac1) and 5 (3CLpro/Mpro) of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2021, 19, 4868–4883. [Google Scholar] [CrossRef]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The role of the nsp2 and nsp3 in its pathogenesis. J. Med. Virol. 2020, 92, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Laamarti, M.; Alouane, T.; Kartti, S.; Chemao-Elfihri, M.; Hakmi, M.; Essabbar, A.; Laamarti, M.; Hlali, H.; Bendani, H.; Boumajdi, N. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. PLoS ONE 2020, 15, e0240345. [Google Scholar] [CrossRef] [PubMed]
- Frick, D.N.; Virdi, R.S.; Vuksanovic, N.; Dahal, N.; Silvaggi, N.R. Molecular basis for ADP-ribose binding to the Mac1 domain of SARS-CoV-2 nsp3. Biochemistry 2020, 59, 2608–2615. [Google Scholar] [CrossRef] [PubMed]
- Virdi, R.S.; Bavisotto, R.V.; Hopper, N.C.; Vuksanovic, N.; Melkonian, T.R.; Silvaggi, N.R.; Frick, D.N. Discovery of drug-like ligands for the Mac1 domain of SARS-CoV-2 Nsp3. SLAS DISCOVERY Adv. Sci. Drug Discov. 2020, 25, 1162–1170. [Google Scholar] [CrossRef]
- Alhammad, Y.M.; Kashipathy, M.M.; Roy, A.; Gagné, J.-P.; McDonald, P.; Gao, P.; Nonfoux, L.; Battaile, K.P.; Johnson, D.K.; Holmstrom, E.D. The SARS-CoV-2 conserved macrodomain is a mono-ADP-ribosylhydrolase. J. Virol. 2021, 95, e01920–e01969. [Google Scholar] [CrossRef]
- Lin, M.-H.; Chang, S.-C.; Chiu, Y.-C.; Jiang, B.-C.; Wu, T.-H.; Hsu, C.-H. Structural, biophysical, and biochemical elucidation of the SARS-CoV-2 nonstructural protein 3 macro domain. ACS Infect. Dis. 2020, 6, 2970–2978. [Google Scholar] [CrossRef]
- Saikatendu, K.S.; Joseph, J.S.; Subramanian, V.; Clayton, T.; Griffith, M.; Moy, K.; Velasquez, J.; Neuman, B.W.; Buchmeier, M.J.; Stevens, R.C. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1″-phosphate dephosphorylation by a conserved domain of nsP3. Structure 2005, 13, 1665–1675. [Google Scholar] [CrossRef]
- Corda, D.; Di Girolamo, M. Functional aspects of protein mono-ADP-ribosylation. EMBO J. 2003, 22, 1953–1958. [Google Scholar] [CrossRef]
- Aravind, L.; Zhang, D.; Souza, R.F.d.; Anand, S.; Iyer, L.M. The natural history of ADP-ribosyltransferases and the ADP-ribosylation system. Endog. ADP-Ribosylation 2015, 384, 3–32. [Google Scholar]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host–pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef]
- Kraus, W.L. PARPs and ADP-Ribosylation: 60 Years on; Cold Spring Harbor Lab.: Laurel Hollow, NY, USA, 2020; Volume 34, pp. 251–253. [Google Scholar]
- Qin, W.; Wu, H.-J.; Cao, L.-Q.; Li, H.-J.; He, C.-X.; Zhao, D.; Xing, L.; Li, P.-Q.; Jin, X.; Cao, H.-L. Research progress on PARP14 as a drug target. Front. Pharmacol. 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Goettsch, C.; Sharma, A.; Ricchiuto, P.; Goh, W.W.B.; Halu, A.; Yamada, I.; Yoshida, H.; Hara, T.; Wei, M. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat. Commun. 2016, 7, 12849. [Google Scholar] [CrossRef]
- Schweiker, S.S.; Tauber, A.L.; Sherry, M.E.; Levonis, S.M. Structure, function and inhibition of poly (ADP-ribose) polymerase, member 14 (PARP14). Mini Rev. Med. Chem. 2018, 18, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Tauber, A.L.; Schweiker, S.S.; Levonis, S.M. The potential association between PARP14 and SARS-CoV-2 infection (COVID-19). Future Med. Chem. 2021, 13, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Michalska, K.; Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Stols, L.; Endres, M.; Joachimiak, A. Crystal structures of SARS-CoV-2 ADP-ribose phosphatase: From the apo form to ligand complexes. IUCrJ 2020, 7, 814–824. [Google Scholar] [CrossRef]
- Tsika, A.C.; Fourkiotis, N.K.; Charalampous, P.; Gallo, A.; Spyroulias, G.A. NMR study of macro domains (MDs) from betacoronavirus: Backbone resonance assignments of SARS–CoV and MERS–CoV MDs in the free and the ADPr-bound state. Biomol. NMR Assign. 2022, 16, 9–16. [Google Scholar] [CrossRef]
- Sherrill, L.M.; Joya, E.E.; Walker, A.; Roy, A.; Alhammad, Y.M.; Atobatele, M.; Wazir, S.; Abbas, G.; Keane, P.; Zhuo, J. Design, synthesis and evaluation of inhibitors of the SARS-CoV-2 nsp3 macrodomain. Bioorg. Med. Chem. 2022, 67, 116788. [Google Scholar] [CrossRef]
- Correy, G.J.; Kneller, D.W.; Phillips, G.; Pant, S.; Russi, S.; Cohen, A.E.; Meigs, G.; Holton, J.M.; Gahbauer, S.; Thompson, M.C. The mechanisms of catalysis and ligand binding for the SARS-CoV-2 NSP3 macrodomain from neutron and X-ray diffraction at room temperature. Sci. Adv. 2022, 8, eabo5083. [Google Scholar] [CrossRef]
- Dasovich, M.; Zhuo, J.; Goodman, J.A.; Thomas, A.; McPherson, R.L.; Jayabalan, A.K.; Busa, V.F.; Cheng, S.-J.; Murphy, B.A.; Redinger, K.R. High-throughput activity assay for screening inhibitors of the SARS-CoV-2 Mac1 Macrodomain. ACS Chem. Biol. 2021, 17, 17–23. [Google Scholar] [CrossRef]
- Brosey, C.A.; Houl, J.H.; Katsonis, P.; Balapiti-Modarage, L.P.; Bommagani, S.; Arvai, A.; Moiani, D.; Bacolla, A.; Link, T.; Warden, L.S. Targeting SARS-CoV-2 Nsp3 macrodomain structure with insights from human poly (ADP-ribose) glycohydrolase (PARG) structures with inhibitors. Prog. Biophys. Mol. Biol. 2021, 163, 171–186. [Google Scholar] [CrossRef]
- Selvaraj, C.; Dinesh, D.C.; Panwar, U.; Boura, E.; Singh, S.K. High-throughput screening and quantum mechanics for identifying potent inhibitors against Mac1 Domain of SARS-CoV-2 Nsp3. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 18, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Schuller, M.; Correy, G.J.; Gahbauer, S.; Fearon, D.; Wu, T.; Díaz, R.E.; Young, I.D.; Carvalho Martins, L.; Smith, D.H.; Schulze-Gahmen, U. Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking. Sci. Adv. 2021, 7, eabf8711. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; McDowell, R.S.; O’Brien, T. Fragment-based drug discovery. J. Med. Chem. 2004, 47, 3463–3482. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, J.B.; Abdolmaleki, A.; Shiri, F. Molecular docking challenges and limitations. In Pharmaceutical Sciences: Breakthroughs in Research and Practice; IGI Global: Hershey, PA, USA, 2017; pp. 770–794. [Google Scholar]
- Singh, D.; Yi, S.V. On the origin and evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef]
- Otto, S.P.; Day, T.; Arino, J.; Colijn, C.; Dushoff, J.; Li, M.; Mechai, S.; Van Domselaar, G.; Wu, J.; Earn, D.J. The origins and potential future of SARS-CoV-2 variants of concern in the evolving COVID-19 pandemic. Curr. Biol. 2021, 31, R918–R929. [Google Scholar] [CrossRef]
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and genomic distribution of SARS-CoV-2 mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic variants of SARS-CoV-2—What do they mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations strengthened SARS-CoV-2 infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Hossain, M.U.; Bhattacharjee, A.; Emon, M.; Hossain, T.; Chowdhury, Z.M.; Ahammad, I.; Mosaib, M.; Moniruzzaman, M.; Rahman, M.; Islam, M. Novel mutations in NSP-1 and PLPro of SARS-CoV-2 NIB-1 genome mount for effective therapeutics. J. Genet. Eng. Biotechnol. 2021, 19, 52. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Parihar, R.D.; Dhiman, U.; Dhamija, P.; Upadhyay, S.K.; Imran, M.; Behera, S.K.; Prasad, T.K. Docking of fda approved drugs targeting nsp-16, n-protein and main protease of SARS-CoV-2 as dual inhibitors. Biointerface Res. Appl. Chem. 2020, 11, 9848–9861. [Google Scholar]
- Prajapat, M.; Sarma, P.; Shekhar, N.; Prakash, A.; Avti, P.; Bhattacharyya, A.; Kaur, H.; Kumar, S.; Bansal, S.; Sharma, A.R. Update on the target structures of SARS-CoV-2: A systematic review. Indian J. Pharmacol. 2020, 52, 142. [Google Scholar] [PubMed]
- Claverie, J.-M. A putative role of de-mono-ADP-ribosylation of STAT1 by the SARS-CoV-2 Nsp3 protein in the cytokine storm syndrome of COVID-19. Viruses 2020, 12, 646. [Google Scholar] [CrossRef]
- Min, W.; Wang, Z.-Q. Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front. Biosci.-Landmark 2009, 14, 1619–1626. [Google Scholar] [CrossRef]
- Patel, D.C.; Hausman, K.R.; Arba, M.; Tran, A.; Lakernick, P.M.; Wu, C. Novel inhibitors to ADP ribose phosphatase of SARS-CoV-2 identified by structure-based high throughput virtual screening and molecular dynamics simulations. Comput. Biol. Med. 2022, 140, 105084. [Google Scholar] [CrossRef]
- Dai, M.; Liu, D.; Liu, M.; Zhou, F.; Li, G.; Chen, Z.; Zhang, Z.; You, H.; Wu, M.; Zheng, Q. Patients with Cancer Appear More Vulnerable to SARS-CoV-2: A Multicenter Study during the COVID-19 OutbreakPatients with Cancer in SARS-CoV-2 Infection. Cancer Discov. 2020, 10, 783–791. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Brogi, S.; Fiorillo, A.; Chemi, G.; Butini, S.; Lalle, M.; Ilari, A.; Gemma, S.; Campiani, G. Structural characterization of Giardia duodenalis thioredoxin reductase (gTrxR) and computational analysis of its interaction with NBDHEX. Eur. J. Med. Chem. 2017, 135, 479–490. [Google Scholar] [CrossRef]
- Artese, A.; Cross, S.; Costa, G.; Distinto, S.; Parrotta, L.; Alcaro, S.; Ortuso, F.; Cruciani, G. Molecular interaction fields in drug discovery: Recent advances and future perspectives. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 594–613. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. In Molecular Modeling of Proteins; Springer: Berlin/Heidelberg, Germany, 2008; pp. 365–382. [Google Scholar]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef]
- Battah, B.; Chemi, G.; Butini, S.; Campiani, G.; Brogi, S.; Delogu, G.; Gemma, S. A Repurposing Approach for Uncovering the Anti-Tubercular Activity of FDA-Approved Drugs with Potential Multi-Targeting Profiles. Molecules 2019, 24, 4373. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; Azevedo, W.F.d. Molegro virtual docker for docking. In Docking Screens for Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2019; pp. 149–167. [Google Scholar]
- Reddy, A.S.; Pati, S.P.; Kumar, P.P.; Pradeep, H.; Sastry, G.N. Virtual screening in drug discovery-a computational perspective. Curr. Protein Pept. Sci. 2007, 8, 329–351. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Norinder, U.; Bergström, C.A. Prediction of ADMET properties. ChemMedChem Chem. Enabling Drug Discov. 2006, 1, 920–937. [Google Scholar]
- Walters, W.P. Going further than Lipinski’s rule in drug design. Expert Opin. Drug Discov. 2012, 7, 99–107. [Google Scholar] [CrossRef]
- Zhang, M.-Q.; Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007, 18, 478–488. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Bakchi, B.; Krishna, A.D.; Sreecharan, E.; Ganesh, V.B.J.; Niharika, M.; Maharshi, S.; Puttagunta, S.B.; Sigalapalli, D.K.; Bhandare, R.R.; Shaik, A.B. An Overview on Applications of SwissADME Web Tool in the Design and Development of Anticancer, Antitubercular and Antimicrobial agents: A Medicinal Chemist’s Perspective. J. Mol. Struct. 2022, 1259, 132712. [Google Scholar] [CrossRef]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. AABC 2015, 8, 37. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Singh, S.; Baker, Q.B.; Singh, D.B. Molecular docking and molecular dynamics simulation. In Bioinformatics; Elsevier: Amsterdam, The Netherlands, 2022; pp. 291–304. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Sirous, H.; Chemi, G.; Gemma, S.; Butini, S.; Debyser, Z.; Christ, F.; Saghaie, L.; Brogi, S.; Fassihi, A.; Campiani, G.; et al. Identification of Novel 3-Hydroxy-pyran-4-One Derivatives as Potent HIV-1 Integrase Inhibitors Using in silico Structure-Based Combinatorial Library Design Approach. Front. Chem. 2019, 7, 574. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | MolDock Score | Re-Rank Score | H-Bond |
|---|---|---|---|
| NSC-358078 | −262.398 | −193.852 | −25.305 |
| NSC-287067 | −242.883 | −186.814 | −9.8173 |
| NSC-353057 | −275.252 | −178.899 | −20.004 |
| NSC-123472 | −236.761 | −178.751 | −5.5102 |
| NSC-20272 | −241.454 | −176.795 | −21.878 |
| NSC-170987 | −252.206 | −175.426 | −21.41 |
| NSC-694613 | −256.587 | −174.499 | −16.771 |
| NSC-729650 | −227.151 | −174.234 | −16.103 |
| NSC-142843 | −214.877 | −172.67 | −20.962 |
| NSC-338956 | −231.941 | −169.875 | −13.263 |
| Compound ID | H-Bond Donor | H-Bond Acceptor | Molecular Weight (g/mol) | LogP | LogS | TPSA (Å2) | Bioavailability Score |
|---|---|---|---|---|---|---|---|
| NSC-358078 | 9 | 19 | 686.48 g/mol | −4.57 | 1.96 | 378.37 | 0.11 |
| NSC-287067 | 2 | 9 | 550.60 g/mol | 3.73 | −5.57 | 112.91 | 0.55 |
| NSC-353057 | 8 | 19 | 778.71 g/mol | −1.47 | −3.1 | 286.89 | 0.11 |
| NSC-123472 | 6 | 12 | 719.55 g/mol | 3.29 | −5.7 | 262.91 | 0.17 |
| NSC-20272 | 7 | 17 | 663.43 g/mol | −4.87 | 0.25 | 340.71 | 0.11 |
| NSC-170987 | 9 | 16 | 626.56 g/mol | −1.06 | −3.16 | 254.52 | 0.17 |
| NSC-694613 | 7 | 11 | 587.68 g/mol | −0.9 | −0.96 | 250.88 | 0.11 |
| NSC-729650 | 8 | 15 | 622.57 g/mol | −0.53 | −2.98 | 234.29 | 0.17 |
| NSC-142843 | 5 | 10 | 483.43 g/mol | −0.22 | −1.61 | 221.56 | 0.11 |
| NSC-338956 | 5 | 14 | 675.71 g/mol | 0.85 | −3.07 | 278.18 | 0.17 |
| Compound ID | ΔGvdw a (kcal/mol) | ΔGcoul b (kcal/mol) | ΔGHbond c (kcal/mol) | ΔGLipo d (kcal/mol) | ΔGPack e (kcal/mol) | ΔGSolGB f (kcal/mol) | ΔGbind g (kcal/mol) |
|---|---|---|---|---|---|---|---|
| NSC-358078 | −71.89 | −37.66 | −7.09 | −19.83 | −1.69 | 35.11 | −89.23 |
| NSC-287067 | −67.12 | −26.19 | −3.12 | −37.76 | −1.35 | 28.83 | −86.75 |
| NSC-142843 | −66.43 | −32.83 | −5.58 | −21.48 | −1.22 | 34.03 | −83.74 |
| NSC-123472 | −64.11 | −30.91 | −3.91 | −24.59 | −0.98 | 31.27 | −81.92 |
| NSC-170987 | −61.51 | −31.37 | −5.71 | −23.38 | −1.10 | 33.94 | −77.31 |
| NSC-20272 | −63.79 | 120.58 | −8.16 | −22.15 | −1.88 | −123.53 | −76.85 |
| NSC-729650 | −55.92 | −22.63 | −3.49 | −38.06 | −1.49 | 31.78 | −69.46 |
| NSC-338956 | −60.89 | −32.86 | −5.01 | −16.17 | −0.64 | 39.29 | −63.56 |
| NSC-353057 | −45.14 | −34.90 | −4.19 | −31.89 | −0.43 | 45.36 | −54.57 |
| NSC-694613 | −40.74 | −13.02 | −4.27 | −20.25 | 0.00 | 19.95 | −48.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yazdani, B.; Sirous, H.; Brogi, S.; Calderone, V. Structure-Based High-Throughput Virtual Screening and Molecular Dynamics Simulation for the Discovery of Novel SARS-CoV-2 NSP3 Mac1 Domain Inhibitors. Viruses 2023, 15, 2291. https://doi.org/10.3390/v15122291
Yazdani B, Sirous H, Brogi S, Calderone V. Structure-Based High-Throughput Virtual Screening and Molecular Dynamics Simulation for the Discovery of Novel SARS-CoV-2 NSP3 Mac1 Domain Inhibitors. Viruses. 2023; 15(12):2291. https://doi.org/10.3390/v15122291
Chicago/Turabian StyleYazdani, Behnaz, Hajar Sirous, Simone Brogi, and Vincenzo Calderone. 2023. "Structure-Based High-Throughput Virtual Screening and Molecular Dynamics Simulation for the Discovery of Novel SARS-CoV-2 NSP3 Mac1 Domain Inhibitors" Viruses 15, no. 12: 2291. https://doi.org/10.3390/v15122291