
Automated SSHHPS Analysis Predicts a Potential Host Protein Target Common to Several Neuroinvasive (+)ssRNA Viruses
 ,
,
Abstract
:
1. Introduction
2. Materials and Methods for In Vitro Analysis and Verification
2.1. Materials
2.2. CFP-YFP Substrate Cloning
2.3. CFP-YFP Substrate Expression and Purification
2.4. In Vitro Protease Assays
2.5. Mass Spectrometry
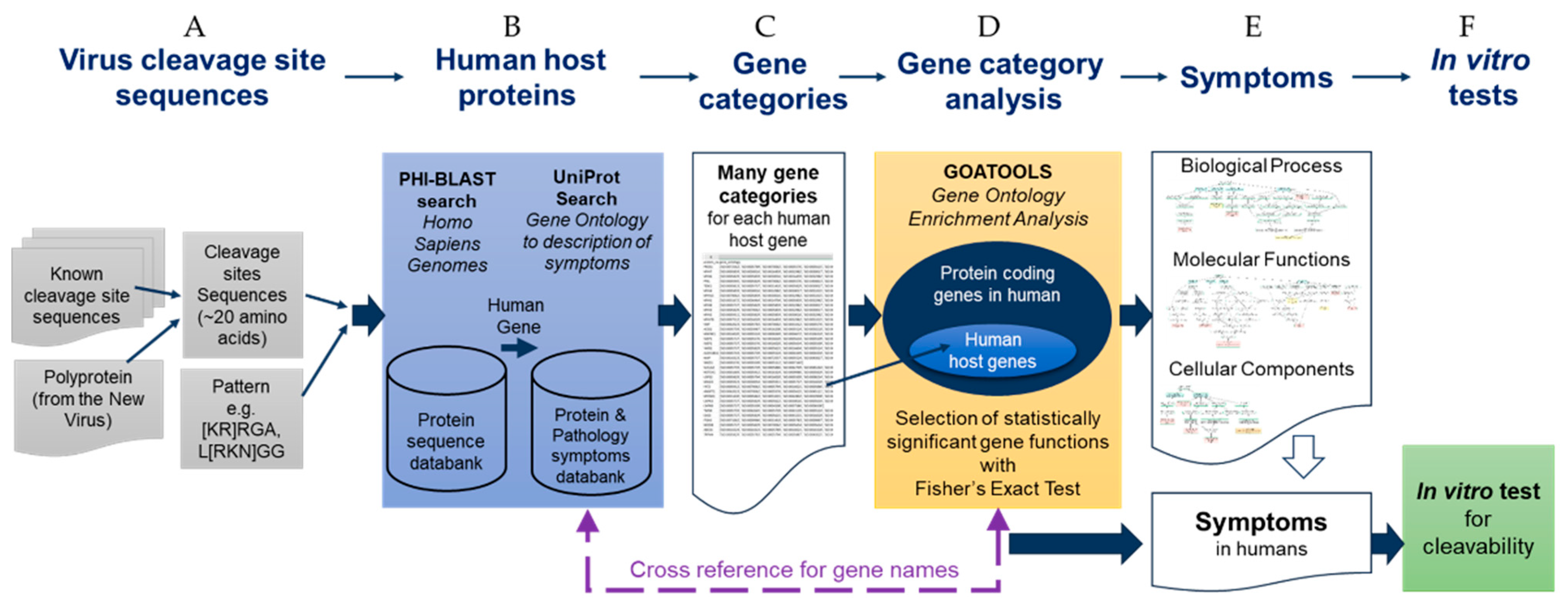
3. Automation Process of the SSHHPS Analysis: The Sequence-To-Symptom Tool
3.1. SSHHPS Analysis of Human Proteins
3.2. Gene Ontology
3.3. Gene Category Analysis
3.3.1. Gene Ontology Enrichment
3.3.2. Fisher’s Exact Test
4. Results
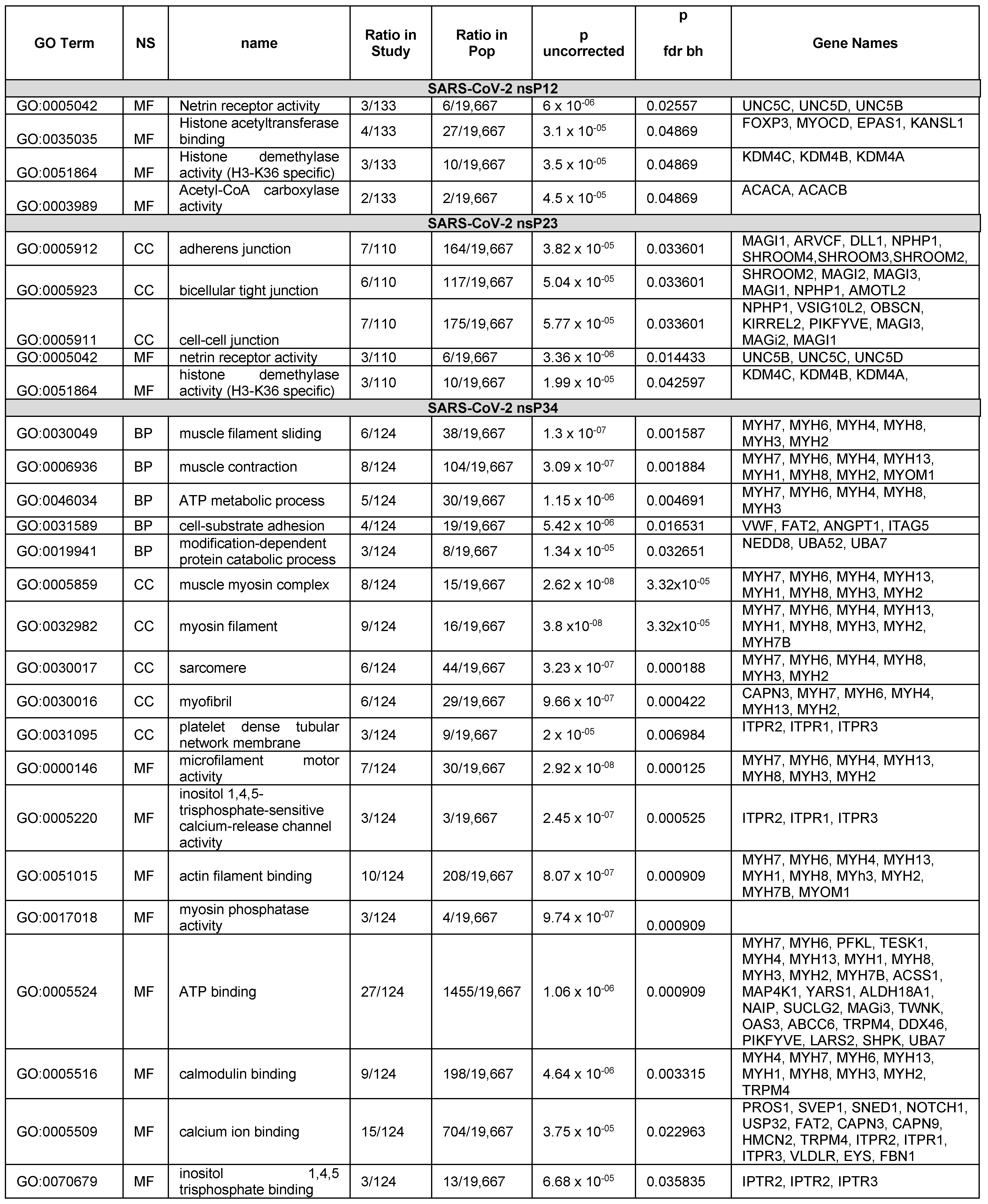
4.1. Gene Ontology Enrichment–Analysis Results
4.1.1. Analysis of the Hits from the SARS-CoV-2 PLpro nsP1/2 Cleavage Site
4.1.2. Analysis of the Hits from the SARS-CoV-2 PLpro nsP2/3 Cleavage Site
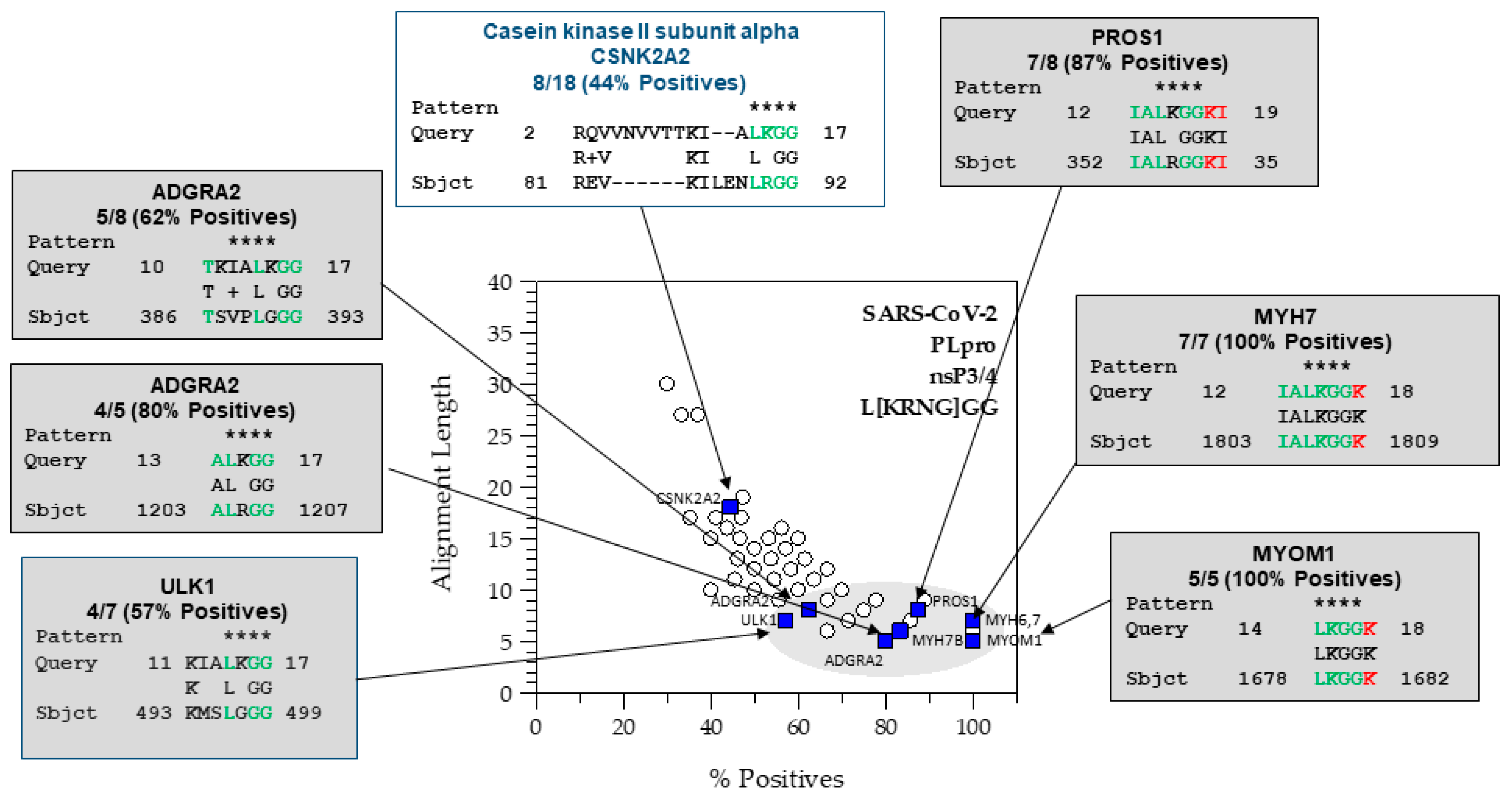
4.1.3. Analysis of the Hits from the SARS-CoV-2 PLpro nsP3/4 Cleavage Site
4.2. In Vitro Cleavage of POT1, VWF, MYOM1, and DNAH8 by the SARS-CoV-2 or MERS PLpro
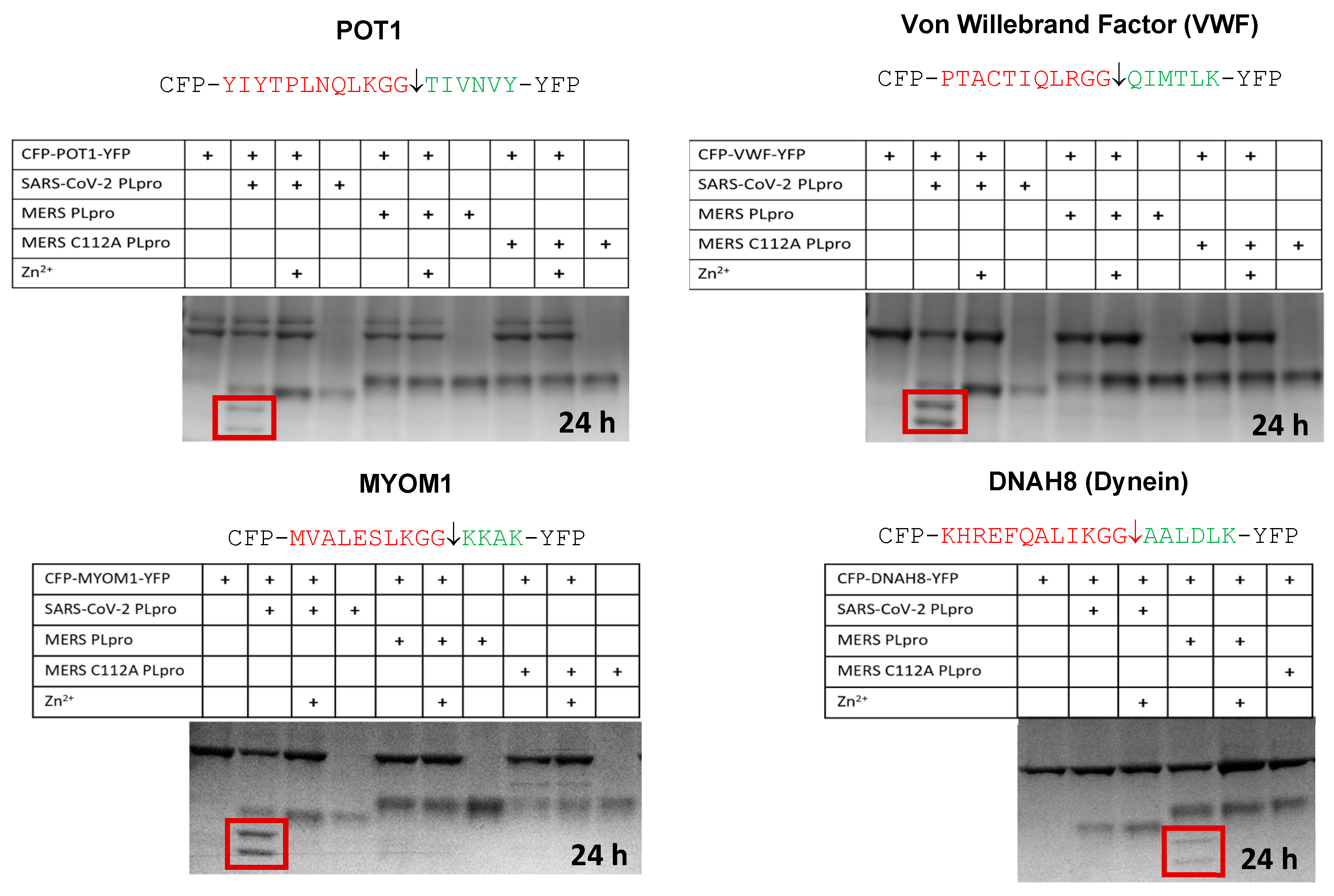

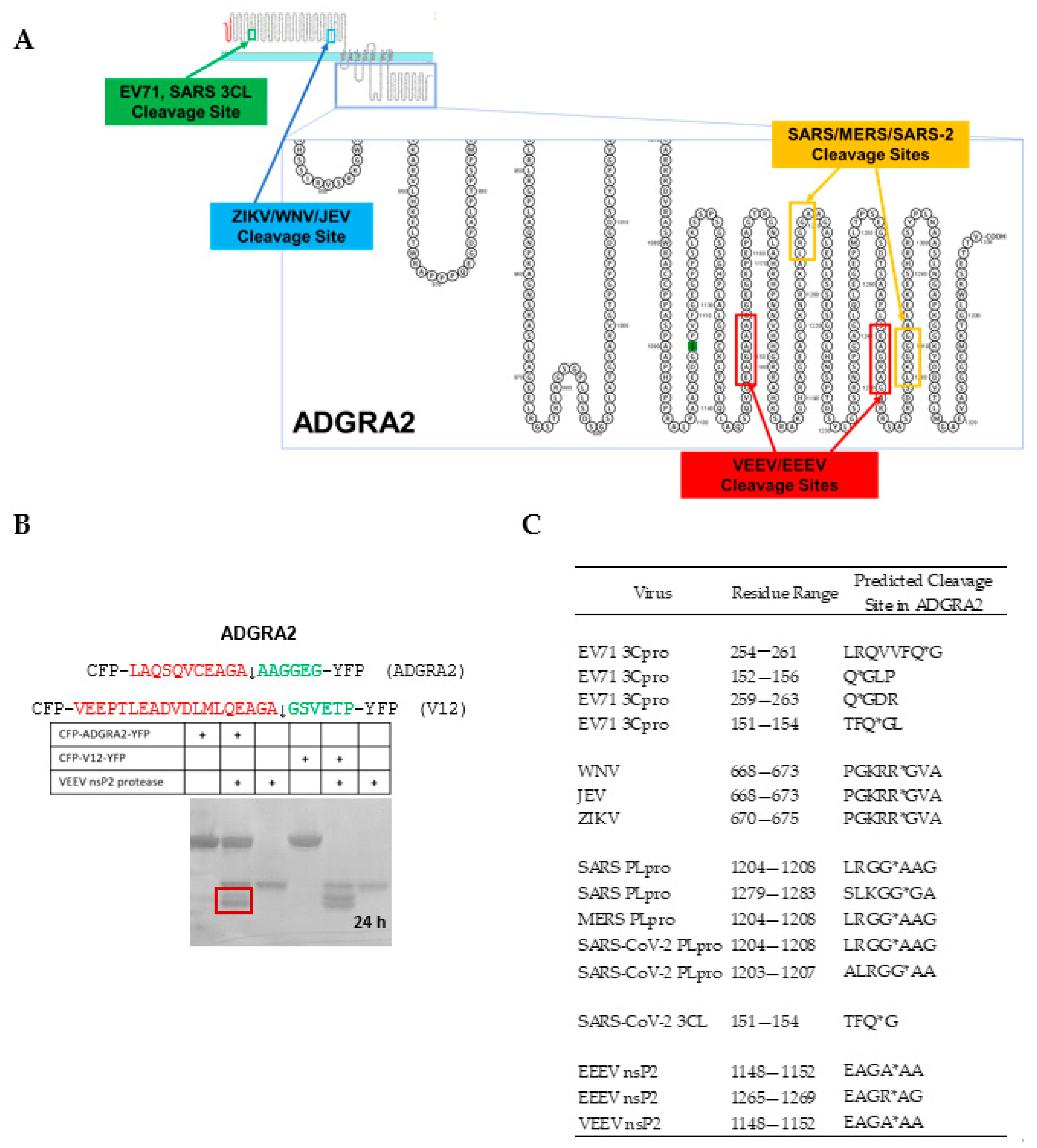
4.3. Cleavage of an ADGRA2 Sequence by the Alphaviral VEEV nsP2 Protease
4.4. Identification of Previously Identified Hits by SSHHPS Analysis
5. Discussion
6. Conclusions
7. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morazzani, E.M.; Compton, J.R.; Leary, D.H.; Berry, A.V.; Hu, X.; Marugan, J.; Glass, P.J.; Legler, P.M. Proteolytic cleavage of host proteins by the Group IV viral proteases of Venezuelan equine encephalitis virus and Zika virus. Antivir. Res. 2019, 164, 106–122. [Google Scholar] [CrossRef]
- Reynolds, N.D.; Aceves, N.M.; Liu, J.L.; Compton, J.R.; Leary, D.H.; Freitas, B.T.; Pegan, S.D.; Doctor, K.Z.; Wu, F.Y.; Hu, X.; et al. The SARS-CoV-2 SSHHPS Recognized by the Papain-like Protease. ACS Infect. Dis. 2021, 7, 1483–1502. [Google Scholar] [CrossRef]
- Hu, X.; Compton, J.R.; Legler, P.M. Analysis of Group IV Viral SSHHPS Using In Vitro and In Silico Methods. J. Vis. Exp. JoVE 2019, 154, e60421. [Google Scholar] [CrossRef]
- Kirkegaard, K.; Baltimore, D. The mechanism of RNA recombination in poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Gorbalenya, A.E. Host-related sequences in RNA viral genomes. Semin. Virol. 1992, 3, 359–371. [Google Scholar]
- Falk, M.M.; Grigera, P.R.; Bergmann, I.E.; Zibert, A.; Multhaup, G.; Beck, E. Foot-and-mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3. J. Virol. 1990, 64, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Grigera, P.R.; Tisminetzky, S.G. Histone H3 modification in BHK cells infected with foot-and-mouth disease virus. Virology 1984, 136, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.M.C. Genetic Recombination in RNA Viruses. In Genetic Diversity of RNA Viruses; Holland, J.J., Ed.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 21–32. [Google Scholar]
- Lytras, S.; Hughes, J.; Martin, D.; Swanepoel, P.; de Klerk, A.; Lourens, R.; Kosakovsky Pond, S.L.; Xia, W.; Jiang, X.; Robertson, D.L. Exploring the Natural Origins of SARS-CoV-2 in the Light of Recombination. Genome Biol. Evol. 2022, 14, evac018. [Google Scholar] [CrossRef] [PubMed]
- Copper, P.D.; Steiner-Pryor, A.; Scotti, P.D.; Delong, D. On the nature of poliovirus genetic recombinants. J. Gen. Virol. 1974, 23, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Gallei, A.; Pankraz, A.; Thiel, H.-J.C.; Becher, P. RNA recombination in vivo in the absence of viral replication. J. Virol. 2004, 78, 6271–6281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, N.; Hansen, J.; Blaas, D.; Brunak, S. Cleavage site analysis in picornaviral polyproteins: Discovering cellular targets by neural networks. Protein Sci. 1996, 5, 2203–2216. [Google Scholar] [CrossRef]
- Kiemer, L.; Lund, O.; Brunak, S.; Blom, N. Coronavirus 3CLpro proteinase cleavage sites: Possible relevance to SARS virus pathology. BMC Bioinform. 2004, 5, 72. [Google Scholar] [CrossRef] [Green Version]
- Scott, B.M.; Lacasse, V.; Blom, D.G.; Tonner, P.D.; Blom, N.S. Predicted coronavirus Nsp5 protease cleavage sites in the human proteome. BMC Genom. Data 2022, 23, 25. [Google Scholar] [CrossRef]
- Miczi, M.; Golda, M.; Kunkli, B.; Nagy, T.; Tőzsér, J.; Mótyán, J.A. Identification of Host Cellular Protein Substrates of SARS-COV-2 Main Protease. Int. J. Mol. Sci. 2020, 21, 9523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; Cheng, G. Protease cleavage of RNF20 facilitates coronavirus replication via stabilization of SREBP1. Proc. Natl. Acad. Sci. USA 2021, 118, e2107108118. [Google Scholar] [CrossRef] [PubMed]
- Badorff, C.; Berkely, N.; Mehrotra, S.; Talhouk, J.W.; Rhoads, R.E.; Knowlton, K.U. Enteroviral protease 2A directly cleaves dystrophin and is inhibited by a dystrophin-based substrate analogue. J. Biol. Chem. 2000, 275, 11191–11197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Bermejo, J.A.; Kang, S.; Rockwood, S.J.; Simoneau, C.R.; Joy, D.A.; Silva, A.C.; Ramadoss, G.N.; Flanigan, W.R.; Fozouni, P.; Li, H.; et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci. Transl. Med. 2008; 13, eabf7872. [Google Scholar]
- Badorff, C.; Knowlton, K.U. Dystrophin disruption in enterovirus-induced myocarditis and dilated cardiomyopathy: From bench to bedside. Med. Microbiol. Immunol. 2004, 193, 121–126. [Google Scholar] [CrossRef]
- Blom, N.; Kreegipuu, A.; Brunak, S.r. PhosphoBase: A database of phosphorylation sites. Nucleic Acids Res. 1998, 26, 382–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stabell, A.C.; Meyerson, N.R.; Gullberg, R.C.; Gilchrist, A.R.; Webb, K.J.; Old, W.M.; Perera, R.; Sawyer, S.L. Dengue viruses cleave STING in humans but not in nonhuman primates, their presumed natural reservoir. Elife 2018, 7, e31919. [Google Scholar] [CrossRef]
- Lim, B.-K.; Peter, A.K.; Xiong, D.; Narezkina, A.; Yung, A.; Dalton, N.D.; Hwang, K.-K.; Yajima, T.; Chen, J.; Knowlton, K.U. Inhibition of Coxsackievirus-associated dystrophin cleavage prevents cardiomyopathy. J. Clin. Investig. 2013, 123, 5146–5151. [Google Scholar] [CrossRef] [Green Version]
- Badorff, C.; Lee, G.-H.; Lamphear, B.J.; Martone, M.E.; Campbell, K.P.; Rhoads, R.E.; Knowlton, K.U. Enteroviral protease 2A cleaves dystrophin: Evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat. Med. 1999, 5, 320–326. [Google Scholar] [CrossRef]
- Cui, S.; Wang, J.; Fan, T.; Qin, B.; Guo, L.; Lei, X.; Wang, J.; Wang, M.; Jin, Q. Crystal Structure of Human Enterovirus 71 3C Protease. J. Mol. Biol. 2011, 408, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T.; Takemoto, C.; Kim, Y.-T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.; Smallwood, P.M.; Nathans, J. Reck and Gpr124 Are Essential Receptor Cofactors for Wnt7a/Wnt7b-Specific Signaling in Mammalian CNS Angiogenesis and Blood-Brain Barrier Regulation. Neuron 2017, 95, 1056–1073.e1055. [Google Scholar] [CrossRef]
- Li, M.Z.; Elledge, S.J. SLIC: A method for sequence- and ligation-independent cloning. Methods Mol. Biol. 2012, 852, 51–59. [Google Scholar] [CrossRef]
- Hu, X.; Compton, J.R.; Leary, D.H.; Olson, M.A.; Lee, M.S.; Cheung, J.; Ye, W.; Ferrer, M.; Southall, N.; Jadhav, A.; et al. Kinetic, Mutational, and Structural Studies of the Venezuelan Equine Encephalitis Virus Nonstructural Protein 2 Cysteine Protease. Biochemistry 2016, 55, 3007–3019. [Google Scholar] [CrossRef] [Green Version]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Wood, V.; Dolinski, K.; Draghici, S. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 2008, 9, 509–515. [Google Scholar] [CrossRef]
- Munoz-Torres, M.; Carbon, S. Get GO! Retrieving GO Data Using AmiGO, QuickGO, API, Files, and Tools. In The Gene Ontology Handbook; Dessimoz, C., Ekunca, N., Eds.; Springer: New York, NY, USA, 2017; pp. 149–160. [Google Scholar]
- Blake, J.A.; Harris, M.A. The Gene Ontology (GO) project: Structured vocabularies for molecular biology and their application to genome and expression analysis. Curr. Protoc. Bioinform. 2008, 23, 7–72. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; RamC-rez, F.; Warwick Vesztrocy, A.; Naldi, A.l.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef]
- Gomez-Mesa, J.E.; Galindo-Coral, S.; Montes, M.C.; MuC1oz Martin, A.s.J. Thrombosis and Coagulopathy in COVID-19. Curr. Probl. Cardiol. 2021, 46, 100742. [Google Scholar] [CrossRef] [PubMed]
- Martin-Rojas, R.M.; Perez-Rus, G.; Delgado-Pinos, V.E.; Domingo-Gonzalez, A.; Regalado-Artamendi, I.; Alba-Urdiales, N.; Demelo-Rodriguez, P.; Monsalvo, S.; Rodriguez-Macias, G.; Ballesteros, M.; et al. COVID-19 coagulopathy: An in-depth analysis of the coagulation system. Eur. J. Haematol. 2020, 105, 741–750. [Google Scholar] [CrossRef]
- Panel, C.-T.G. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. National Institutes of Health. Available online: https://www.covid19treatmentguidelines.nih.gov/ (accessed on 22 August 2022).
- Gildenhuys, S. Expanding our understanding of the role polyprotein conformation plays in the coronavirus life cycle. Biochem. J. 2020, 477, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Lulla, V.; Karo-Astover, L.; Rausalu, K.; Saul, S.; Merits, A.; Lulla, A. Timeliness of Proteolytic Events Is Prerequisite for Efficient Functioning of the Alphaviral Replicase. J. Virol. 2018, 92, e00151-18. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Li, B.; Zhou, Z.; Hancock, W.W.; Zhang, H.; Greene, M.I. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr. Opin. Immunol. 2010, 22, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Mancuso, M.R.; Maier, C.; Liang, X.; Yuki, K.; Yang, L.; Kwong, J.W.; Wang, J.; Rao, V.; Vallon, M.; et al. Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat. Med. 2017, 23, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Omasits, U.; Ahrens, C.H.; Muller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamud, Y.; Xue, Y.C.; Liu, H.; Ng, C.S.; Bahreyni, A.; Jan, E.; Luo, H. The papain-like protease of coronaviruses cleaves ULK1 to disrupt host autophagy. Biochem. Biophys. Res. Commun. 2021, 540, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Moustaqil, M.; Ollivier, E.; Chiu, H.P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microbes Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef]
- Hameedi, M.A.; Prates, E.T.; Garvin, M.R.; Mathews, I.I.; Amos, B.K.; Demerdash, O.; Bechthold, M.; Iyer, M.; Rahighi, S.; Kneller, D.W.; et al. Structural and functional characterization of NEMO cleavage by SARS-CoV-2 3CLpro. Nat. Commun. 2022, 13, 5285. [Google Scholar] [CrossRef]
- Wenzel, J.; Lampe, J.; Müller-Fielitz, H.; Schuster, R.; Zille, M.; Müller, K.; Krohn, M.; Körbelin, J.; Zhang, L.; Özorhan, C.; et al. The SARS-CoV-2 main protease M(pro) causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24, 1522–1533. [Google Scholar] [CrossRef]
- Magg, T.; Mannert, J.; Ellwart, J.W.; Schmid, I.; Albert, M.H. Subcellular localization of FOXP3 in human regulatory and nonregulatory T cells. Eur. J. Immunol. 2012, 42, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Baez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Shiryaev, S.A.; Ratnikov, B.I.; Chekanov, A.V.; Sikora, S.; Rozanov, D.V.; Godzik, A.; Wang, J.; Smith, J.W.; Huang, Z.; Lindberg, I.; et al. Cleavage targets and the D-arginine-based inhibitors of the West Nile virus NS3 processing proteinase. Biochem. J. 2006, 393, 503–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balistreri, G.; Horvath, P.; Schweingruber, C.; Zünd, D.; McInerney, G.; Merits, A.; Mühlemann, O.; Azzalin, C.; Helenius, A. The host nonsense-mediated mRNA decay pathway restricts Mammalian RNA virus replication. Cell Host Microbe. 2014, 16, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Kongpracha, P.; Wiriyasermkul, P.; Isozumi, N.; Moriyama, S.; Kanai, Y.; Nagamori, S. Simple But Efficacious Enrichment of Integral Membrane Proteins and Their Interactions for In-Depth Membrane Proteomics. Mol. Cell Proteom. 2022, 21, 100206. [Google Scholar] [CrossRef]
- Martin, M.; Vermeiren, S.; Bostaille, N.; Eubelen, M.; Spitzer, D.; Vermeersch, M.; Profaci, C.P.; Pozuelo, E.; Toussay, X.; Raman-Nair, J.; et al. Engineered Wnt ligands enable blood-brain barrier repair in neurological disorders. Science 2022, 375, eabm4459. [Google Scholar] [CrossRef]
- Legler, P.M.; Morazzani, E.; Glass, P.J. Methods and Compositions for the Detection of Host Protein Cleavage By Group IV Viral Proteases. U.S. Patent Application No. 17/458,812, 27 August 2021. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Scissile Bond Cut by Viral Protease | Examples |
|---|---|---|
| Picornaviridae | Q↓G, Q↓A, Q↓S, Q↓H, Q↓V, Q↓L | Poliovirus, EV68, EV71, Hepatitis A, Foot-and-mouth disease, Coxsackie B virus |
| Calciviridae | Q↓G, E↓G, E↓A, Q↓T | Norovirus, Feline calicivirus |
| Flaviviridae | RR↓G, KR↓G, RYR↓G, KRR↓(S/T) | JEV (Japanese encephalitisvirus), Dengue, West Nile, Zika (ZIKV), Hepatitis C (HepC), Yellow Fever (YFV) |
| Togaviridae | QEAGA↓G, HEAG(CR)↓A, (FY)(DE)AGA↓Y, (GA)(GA)↓G | Alphaviruses (VEEV/EEEV/WEEV, Sindbis, SFV, Chikungunya, Ross River), Rubella |
| Coronaviridae | LKGG↓, LRGG↓, LNGG↓ (PLpro deUb/deISGylase) Q↓G, Q↓S, Q↓A, Q↓N (3CL Mpro) | SARS, MERS, SARS-2 |
| SARS-CoV-2 PLpro | ||||||
|---|---|---|---|---|---|---|
| Align. | nsp1/2 | SGVTRELMRELNGG↓AYTRYV | Cleavage | Host protein target containing | ||
| Length | %Pos | nsp2/3 | NMMVTNNTFTLKGG↓APTKVT | In Vitro | a predicted cleavage site | Ref. |
| (aa) | nsp3/4 | VVNVVTTKIALKGG↓KIVNNW | ||||
| 8 | 88% | LLIALRGG↓KIEVQL | Y | Vit. K-dep. Protein S (PROS1) | [2] | |
| 7 | 100% | EAEQIALKGG↓KKQLQK | Y | MYH7, cardiac myosin | [2] | |
| 7 | 100% | EAEQIALKGG↓KKQLQK | Y | MYH6, cardiac myosin | [2] | |
| 8 | 88% | PLNQLKGG↓TIVNVY | Y | Protection of telomeres 1 (POT1) | (this paper) | |
| 7 | 86% | FQGRDLRGG↓AHASSS | Y | FOXP3 | [2] | |
| 6 | 83% | GYCFRELRGG|ECASPL | N | LTBP4 | ||
| 6 | 83% | NLTEILNGG↓VYVDQN | Y | ErbB4 (HER4) | [2] | |
| 8 | 63% | ACTIQLRGG↓QIMTLK | Y | VWF | (this paper) | |
| 5 | 100% | RMAALESLKGG↓KKAK- | Y | Myomesin-1 (MYOM1) | (this paper) | |
| MERS PLpro | ||||||
| 5 | 80% | KHREFQALIKGG↓AALDLK | Y | DNAH8 (dynein axonemal heavy chain 8) | (this paper) | |
| VEEV nsP2 Protease | ||||||
| Align. | nsP1/2 | VEEPTLEADVDLMLQEAGA↓GSVETP | Y | |||
| Length | %Pos | nsP2/3 | LSSTLTNIYTGSRLHEAGC↓APSYHV | Y | ||
| (aa) | nsP3/4 | TREEFEAFVAQQQRFDAGA↓YIFSSD | Y | |||
| 5 | 80% | LAQSQVCEAGA↓AAGGEG | Y | ADGRA2 | (this paper) | |
| 5 | 80% | DCFATGRHYWEVDVQEAGA↓GWWVGA | Y | TRIM14 | [1] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doctor, K.Z.; Gilmour, E.; Recarte, M.; Beatty, T.R.; Shifa, I.; Stangel, M.; Schwisow, J.; Leary, D.H.; Legler, P.M. Automated SSHHPS Analysis Predicts a Potential Host Protein Target Common to Several Neuroinvasive (+)ssRNA Viruses. Viruses 2023, 15, 542. https://doi.org/10.3390/v15020542
Doctor KZ, Gilmour E, Recarte M, Beatty TR, Shifa I, Stangel M, Schwisow J, Leary DH, Legler PM. Automated SSHHPS Analysis Predicts a Potential Host Protein Target Common to Several Neuroinvasive (+)ssRNA Viruses. Viruses. 2023; 15(2):542. https://doi.org/10.3390/v15020542
Chicago/Turabian StyleDoctor, Katarina Z., Elizabeth Gilmour, Marilyn Recarte, Trinity R. Beatty, Intisar Shifa, Michaela Stangel, Jacob Schwisow, Dagmar H. Leary, and Patricia M. Legler. 2023. "Automated SSHHPS Analysis Predicts a Potential Host Protein Target Common to Several Neuroinvasive (+)ssRNA Viruses" Viruses 15, no. 2: 542. https://doi.org/10.3390/v15020542