
Molecular Epidemiology of SARS-CoV-2 during Five COVID-19 Waves and the Significance of Low-Frequency Lineages

 , , , , , , ,
, , , , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Samples
2.2. Next-Generation Sequencing of SARS-CoV-2 Strains
2.3. Data Analysis
3. Results
3.1. Study Population Demographics
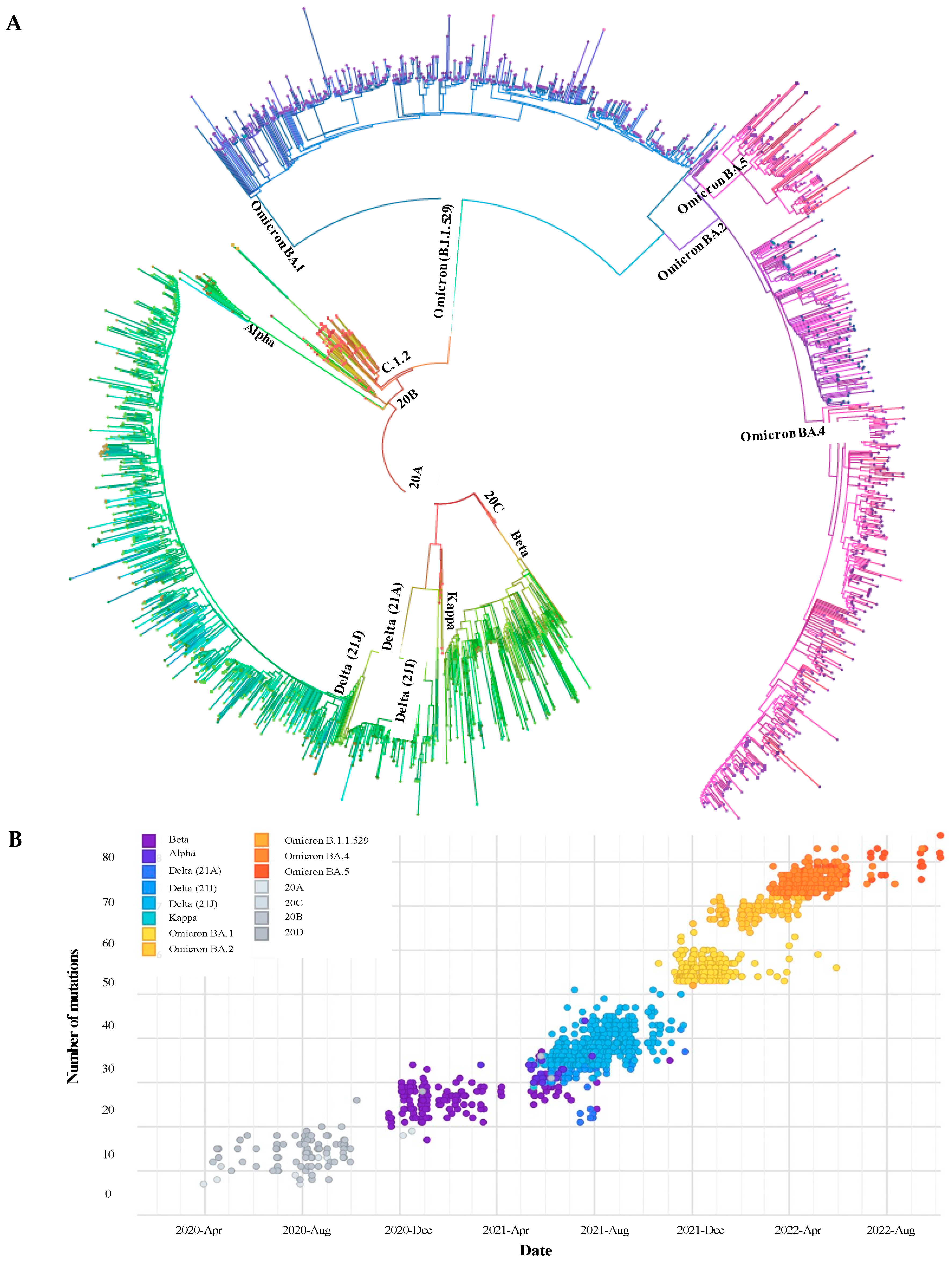
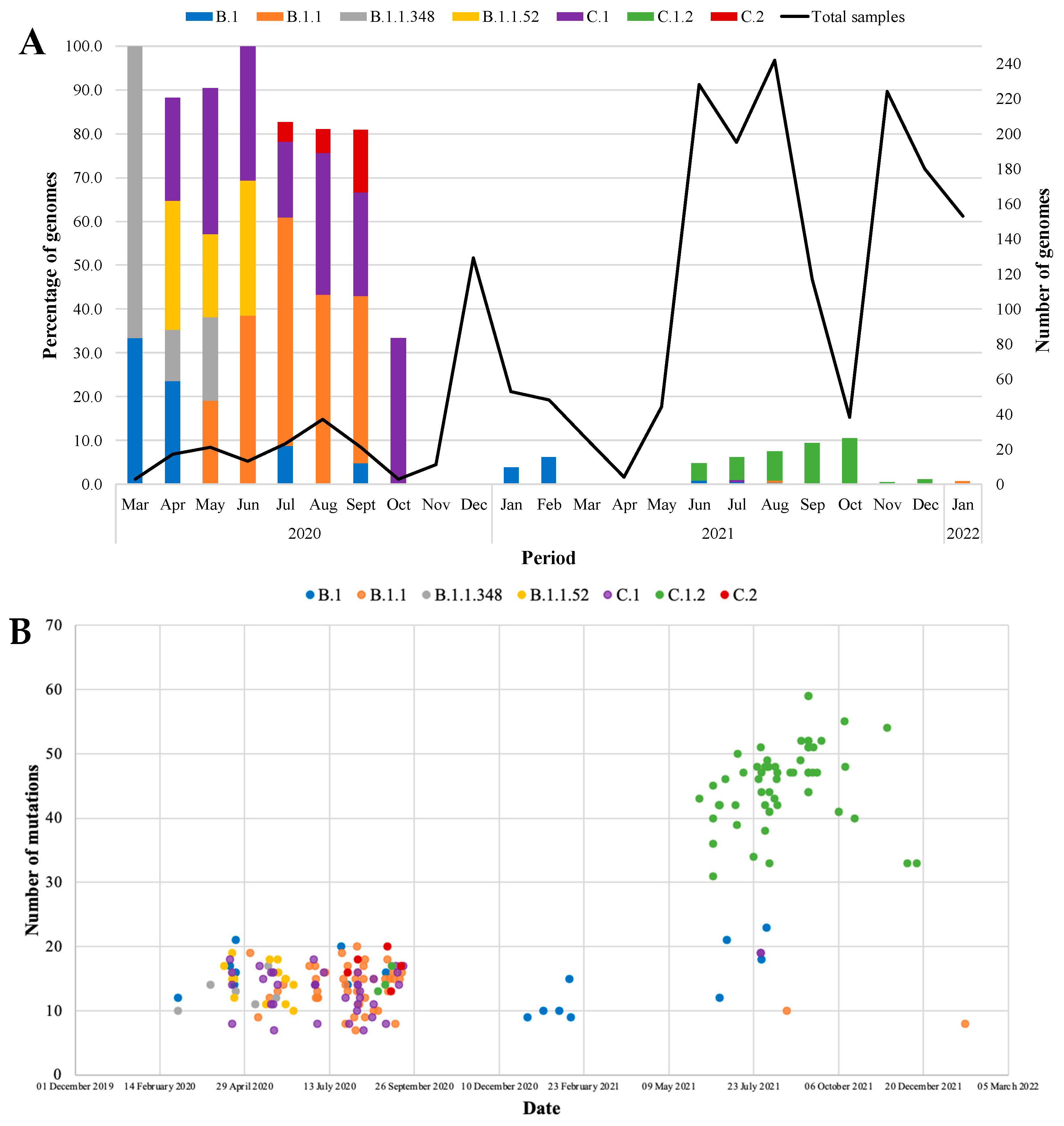
3.2. Epidemiology of SARS-CoV-2 Lineages and VOCs, 2020 to 2022
3.3. Phylogenetic Description of VOCs Overtime
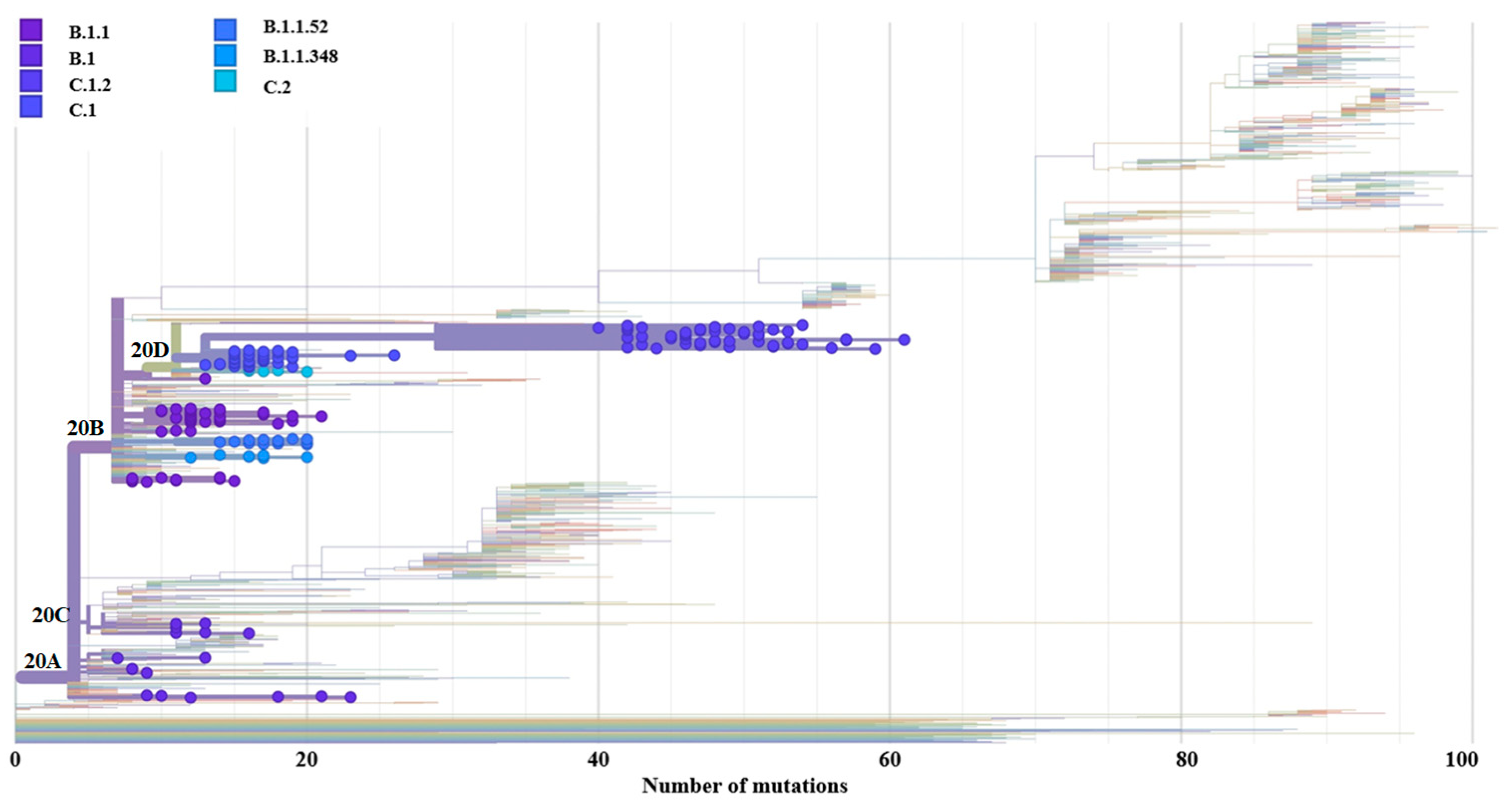
3.4. Genome-Wide Diversity of Low-Frequency Lineages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; de Silva, T.I.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Wilkinson, E.; Giovanetti, M.; Tegally, H.; San, J.; Lessels, R.; Cuadros, D.; Martin, D.; Zekri, A.-R.; Sangare, A.; Ouedraogo, A.-S.; et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science 2021, 431, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Dhar, M.S.; Marwal, R.; Radhakrishnan, V.S.; Ponnusamy, K.; Jolly, B.; Bhoyar, R.C.; Sardana, V.; Naushin, S.; Rophina, M.; Mellan, T.A.; et al. Genomic characterization and epidemiology of an emerging SARS-CoV-2 variant in Delhi, India. Science 2021, 374, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Moyo, S.; Amoako, D.G.; Althaus, C.L. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1789. [Google Scholar] [CrossRef]
- Department of Health Republic of South Africa. COVID-19 Policies. Available online: https://www.health.gov.za/covid19/downloads.html?type=policies%0D%0A (accessed on 10 May 2023).
- SAPHRA (South African Health Products Regulatory Authority). Available online: https://www.sahpra.org.za/news-and-updates/sahpra-authorises-covishield-an-adenovirus-vectored-vaccine-for-the-prevention-of-covid-19/ (accessed on 10 May 2023).
- World Health Organisation. Available online: https://covid19.who.int/ (accessed on 10 May 2023).
- GISAID. Available online: https://www.epicov.org/epi3/frontend#30823%0D%0A (accessed on 10 May 2023).
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S.; et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Scheepers, C.; Everatt, J.; Amoako, D.G.; Tegally, H.; Wibmer, C.K.; Mnguni, A.; Ismail, A.; Mahlangu, B.; Lambson, B.E.; Richardson, S.I.; et al. Emergence and phenotypic characterization of the global SARS-CoV-2 C.1.2 lineage. Nat. Commun. 2022, 13, 1976. [Google Scholar] [CrossRef]
- Brandt, C.; Spott, R.; Hölzer, M.; Kühnert, D.; Fuchs, S.; Lohde, M.; Marquet, M.; Viehweger, A.; Rimek, D.; Pletz, M.W. Molecular epidemiology of SARS-CoV-2—A regional to global perspective. medRxiv 2021, 1–9. [Google Scholar] [CrossRef]
- Quick, J. nCoV-2019 Sequencing Protocol v3 (LoCost) V.3. 2020. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8yeJosh (accessed on 2 November 2022).
- Reiling, S.J.; Loranger, K.; Roy, A.-M.; Chen, S.-H.; Ragoussis, I. nCoV-2019 McGill Artic PCR Protocol, V4.1 at 63C V.2. McGill Genome Centre. 2022. Available online: https://www.protocols.io/view/ncov-2019-mcgill-artic-pcr-protocol-v4-1-at-63c-ewov18e4ygr2/v2 (accessed on 2 November 2022).
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Cleemput, S.; Dumon, W.; Fonseca, V.; Abdool Karim, W.; Giovanetti, M.; Carlos Alcantara, L.; Deforche, K.; de Oliveira, T. Genome Detective Coronavirus Typing Tool for rapid identification and characterization of novel coronavirus genomes. Bioinformatics 2020, 11, 3552–3555. [Google Scholar] [CrossRef] [Green Version]
- Exatype: Automated SARS-CoV-2 Variant Typing Directly from NGS Sequence Data Exatype. Hyrax BioSciences. Available online: https://ngs.exatype.com/dashboard?product=SARSCoV2 (accessed on 2 February 2022).
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021, 590, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.; Lipsitch, M.; Cevik, M. On the effect of age on the transmission of SARS-CoV-2 in households, schools and the community. J. Infect. Dis. 2021, 223, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Cécile, T.K.; Bosetti, P.; Paireau, J.; Crépey, P.; Salje, H.; Lefrancq, N.; Fontanet, A.; Benamouzig, D.; Boëlle, P.Y.; Desenclos, J.C.; et al. SARS-CoV-2 transmission across age groups in France and implications for control. Nat. Commun. 2021, 12, 6895. [Google Scholar] [CrossRef]
- Phaswana-Mafuya, N.; Shisana, O.; Jassat, W.; Baral, S.D.; Makofane, K.; Phalane, E.; Zuma, K.; Zungu, N.; Chadyiwa, M. Understanding the differential impacts of COVID-19 among hospitalised patients in South Africa for equitable response. S. Afr. Med. J. 2021, 111, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Monod, M.; Blenkinsop, A.; Xi, X.; Hebert, D.; Bershan, S.; Tietze, S.; Baguelin, M.; Bradley, V.C.; Chen, Y.; Coupland, H.; et al. Age groups that sustain resurging COVID-19 epidemics in the United States. Science 2021, 371, eabe8372. [Google Scholar] [CrossRef]
- National Institute for Communicable Diseases. SARS-CoV-2 Genomic Surveillance Update June 2021. 2021. Available online: https://www.nicd.ac.za/wp-content/uploads/2021/07/Sequencing-update-1July-2021_V14.pdf (accessed on 2 February 2023).
- Network for Genomic Surveillance in South Africa (NGS-SA). SARS-CoV-2 Sequencing Update 7 July 2021. 2021. Available online: https://www.nicd.ac.za/wp-content/uploads/2021/12/Update-of-SA-sequencing-data-from-GISAID-1-Dec-Final.pdf (accessed on 2 February 2023).
- Gangavarapu, K.; Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K.; et al. Outbreak.info genomic reports: Scalable and dynamic surveillance of SARS-CoV-2 variants 2 and mutations. Nat. Methods 2023, 20, 512–522. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.C.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. Genomic epidemiology of SARS-CoV-2 reveals multiple lineages and early spread of SARS-CoV-2 infections in Lombardy, Italy. Nat. Commun. 2021, 12, 434. [Google Scholar] [CrossRef]
- Andrés, C.; Piñana, M.; Borràs-Bermejo, B.; González-Sánchez, A.; García-Cehic, D.; Esperalba, J.; Rando, A.; Zules-Oña, R.G.; Campos, C.; Codina, M.G.; et al. A year living with SARS-CoV-2: An epidemiological overview of viral lineage circulation by whole-genome sequencing in Barcelona city (Catalonia, Spain). Emerg. Microbes Infect. 2022, 11, 172–181. [Google Scholar] [CrossRef]
- Silva, C.L.J.; Rivero, R.; Douglas, J.; Bouckaert, R.; Arenas, C.J.V.; Atkins, K.; Gastelbondo, B.; Calderon, A.; Guzman, C.; Echeverri-De la Hoz, D.; et al. Genomic epidemiology of circulating SARS-CoV-2 variants during first two years of the pandemic in Colombia. medRxiv 2022, 1–24. [Google Scholar] [CrossRef]
- Quispe-Ricalde, M.A.; Castelán-Sánchez, H.G.; Meza-Rodríguez, P.M.; Dávila-Ramos, S.; Sierra, J.L.; Batista-Garcia, R.; Concha-Velasco, F.; Lucana, S.F.; De Santa Cruz, J.; Zea, V.; et al. Evidence of natural selection and dominance of SARS-CoV-2 variant Lambda (C.37) over variants of concern in Cusco, Peru. Arch. Virol. 2023, 168, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Oróstica, K.Y.; Contreras, S.; Mohr, S.B.; Dehning, J.; Ulloa, S.; Castillo, A.E.; Verdugo, R.A.; Fernández, J. Mutational signatures and transmissibility of SARS-CoV-2 Gamma and Lambda variants. arXiv 2021, arXiv:2108.10018. [Google Scholar] [CrossRef]
- Zambrana Montaño, R.; Culasso, A.C.A.; Fernández, F.; Marquez, N.; Debat, H.; Salmerón, M.; Zamora, A.M.; Ruíz de Huidobro, G.; Costas, D.; Alabarse, G.; et al. Evolution of SARS-CoV-2 during the first year of the COVID-19 pandemic in Northwestern Argentina. Virus Res. 2023, 323, 198936. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Sternberg, Á.M.; Chaparro-Solano, H.M.; Albornóz, L.L.; Pinzón-Rondón, Á.M.; Pardo-Oviedo, J.M.; Molano-González, N.; Otero-Rodríguez, D.A.; Zapata-Gómez, F.A.; Gálvez, J.M. Genomic characterization of SARS-CoV-2 and its association with clinical outcomes: A 1-year longitudinal study of the pandemic in Colombia. Int. J. Infect. Dis. 2022, 116, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, P.; Okendo, J.; Mogotsi, M.; Ogunbayo, A.; Adelabu, O.; Sondlane, H.; Maotoana, M.; Mahomed, L.; Morobadi, M.D.; Vawda, S.; et al. SARS-CoV-2 variants from COVID-19 positive cases in the Free State province, South Africa from July 2020 to December 2021. Front. Virol. 2022, 2, 935131. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Mou, K.; Abdalla, M.; Qing, D.; Tahir, M. Emerging mutations in envelope protein of SARS-CoV-2 and their effect on thermodynamic properties. Inform. Med. Unlocked 2020, 25, 100675. [Google Scholar] [CrossRef]
- Liu, S.; Shen, J.; Fang, S.; Li, K.; Liu, J.; Yang, L.; Hu, C.D.; Wan, J. Genetic Spectrum and Distinct Evolution Patterns of SARS-CoV-2. Front. Microbiol. 2020, 11, 593548. [Google Scholar] [CrossRef]
- Shen, L.; Bard, J.D.; Triche, T.J.; Judkins, A.R.; Biegel, J.A.; Gai, X. Emerging variants of concern in SARS-CoV-2 membrane protein: A highly conserved target with potential pathological and therapeutic implications. Emerg. Microbes Infect. 2021, 10, 885–893. [Google Scholar] [CrossRef]
- Marques-Pereira, C.; Pires, M.N.; Gouveia, R.P.; Pereira, N.N.; Caniceiro, A.B.; Rosário-Ferreira, N.; Moreira, I.S. SARS-CoV-2 Membrane Protein: From Genomic Data to Structural New Insights. Int. J. Mol. Sci. 2022, 23, 2986. [Google Scholar] [CrossRef]
- Xia, S.; Wang, L.; Zhu, Y.; Lu, L.; Jiang, S. Origin, virological features, immune evasion and intervention of SARS-CoV-2 Omicron sublineages. Signal Transduct. Target. Ther. 2022, 7, 241. [Google Scholar] [CrossRef] [PubMed]
- Alsuwairi, F.A.; Alsaleh, A.; Alsanea, M.S.; Al-qahtani, A.A. Association of SARS-CoV-2 Nucleocapsid Protein Mutations with Patient Demographic and Clinical Characteristics during the Delta and Omicron Waves. Microorganisms 2023, 11, 1288. [Google Scholar] [CrossRef]
- Sant’Anna, F.H.; Muterle Varela, A.P.; Prichula, J.; Comerlato, J.; Comerlato, C.B.; Roglio, V.S.; Mendes Pereira, G.F.; Moreno, F.; Seixas, A.; Wendland, E.M. Emergence of the novel SARS-CoV-2 lineage VUI-NP13L and massive spread of P.2 in South Brazil. Emerg. Microbes Infect. 2021, 10, 1431–1440. [Google Scholar] [CrossRef]
- Ghaleh, S.S.; Rahimian, K.; Mahmanzar, M.; Mahdavi, B.; Tokhanbigli, S.; Sisakht, M.M.; Farhadi, A.; Bakhtiari, M.M.; Kuehu, D.L.; Deng, Y. SARS-CoV-2 Non-structural protein 1(NSP1) mutation virulence and natural selection: Evolutionary trends in the six continents. Virus Res. 2023, 323, 199016. [Google Scholar] [CrossRef] [PubMed]
- Moen, L.V.; Vollan, H.S.; Bråte, J.; Hungnes, O.; Bragstad, K. Molecular Epidemiology of the Norwegian SARS-CoV-2 Delta Lineage AY.63. Viruses 2022, 14, 2734. [Google Scholar] [CrossRef] [PubMed]
- Azad, G.K.; Khan, P.K. Variations in Orf3a protein of SARS-CoV-2 alter its structure and function. Biochem. Biophys. Rep. 2021, 26, 100933. [Google Scholar] [CrossRef]
- Eskier, D.; Karakülah, G.; Suner, A.; Oktay, Y. RdRp mutations are associated with SARS-CoV-2 genome evolution. PeerJ 2020, 8, e9587. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Peacock, T.P.; Penrice-Randal, R.; Hiscox, J.A.; Barclay, W.S. SARS-CoV-2 one year on: Evidence for ongoing viral adaptation. J. Gen. Virol. 2021, 102, 001584. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Takano, K.; Watanabe, Y.; Hariu, M.; Seki, M. Detection of representative mutant strains and a case of prolonged infection by SARS-CoV-2 with spike 69/70 deletion in Japan. Infect. Drug Resist. 2021, 14, 2579–2581. [Google Scholar] [CrossRef]
- Chen, K.W.K.; Tsung-Ning Huang, D.; Huang, L.M. SARS-CoV-2 variants—Evolution, spike protein, and vaccines. Biomed. J. 2022, 45, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Zhang, L.; Farzan, M.; Choe, H. Functional importance of the D614G mutation in the SARS-CoV-2 spike protein. Biochem. Biophys. Res. Commun. 2021, 538, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Abavisani, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Mollapour Siasakht, M.; Farhadi, A.; Kodori, M.; Mahmanzar, M.; Meshkat, Z. Mutations in SARS-CoV-2 structural proteins: A global analysis. Virol. J. 2022, 19, 220. [Google Scholar] [CrossRef] [PubMed]
- Candido, K.L.; Eich, C.R.; de Fariña, L.O.; Kadowaki, M.K.; da Conceição Silva, J.L.; Maller, A.; Simão, R.D.C.G. Spike protein of SARS-CoV-2 variants: A brief review and practical implications. Braz. J. Microbiol. 2022, 53, 1133–1157. [Google Scholar] [CrossRef] [PubMed]
- Asif, A.; Ilyas, I.; Abdullah, M.; Sarfraz, S.; Mustafa, M.; Mahmood, A. The Comparison of Mutational Progression in SARS-CoV-2: A Short Updated Overview. J. Mol. Pathol. 2022, 3, 201–218. [Google Scholar] [CrossRef]
- Network for Genomic Surveillance in South Africa (NGS-SA). SARS-CoV-2 Seq Updat 3 December 2021. 2021, pp. 1–29. Available online: https://www.nicd.ac.za/wp-content/uploads/2021/12/Update-of-SA-sequencing-data-from-GISAID-3-Dec-21-Final.pdf (accessed on 2 November 2022).
- Network for Genomic Surveillance in South Africa (NGS-SA). SARS-CoV-2 Seq Update 14 January 2022. Glob Challenges. 2022, pp. 1–32. Available online: https://www.nicd.ac.za/wp-content/uploads/2022/01/Update-of-SA-sequencing-data-from-GISAID-14-Jan-2022_dash_v2-Read-Only.pdf (accessed on 2 November 2022).
- Subramoney, K.; Mtileni, N.; Bharuthram, A.; Davis, A.; Kalenga, B.; Rikhotso, M.; Maphahlele, M.; Giandhari, J.; Naidoo, Y.; Pillay, S.; et al. Identification of SARS-CoV-2 Omicron variant using spike gene target failure and genotyping assays, Gauteng, South Africa, 2021. J. Med. Virol. 2022, 94, 3676–3684. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Althaus, C.L.; Giovanetti, M.; San, J.E.; Giandhari, J.; Pillay, S.; Naidoo, Y.; Ramphal, U.; Msomi, N.; et al. Rapid replacement of the Beta variant by the Delta variant in South Africa. medRxiv 2021, 1–26. [Google Scholar] [CrossRef]
- Gangavarapu, K.; Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K.; et al. Lineage Comparison. 2023. Available online: https://outbreak.info/compare-lineages?pango (accessed on 13 February 2023).
- World Health Organization. COVID-19 Weekly Epidemiological Update Edition 70. Edition 70, Published 14 December 2021. 2021. Available online: https://apps.who.int/iris/handle/10665/350646 (accessed on 30 January 2023).
- Dhawan, M.; Sharma, A.; Priyanka; Thakur, N.; Rajkhowa, T.K.; Choudhary, O.P. Delta variant (B.1.617.2) of SARS-CoV-2: Mutations, impact, challenges and possible solutions. Hum. Vaccines Immunother. 2022, 18, 2068883. [Google Scholar] [CrossRef]
- Mohapatra, R.K.; Kandi, V.; Tuli, H.S.; Chakraborty, C.; Dhama, K. The recombinant variants of SARS-CoV-2: Concerns continues amid COVID-19 pandemic. J. Med. Virol. 2022, 94, 3506–3508. [Google Scholar] [CrossRef]
- World Health Organization. COVID-19 Weekly Epidemiological Update Edition 115. Published 26 October 2022. 2022. Available online: https://apps.who.int/iris/handle/10665/363853 (accessed on 30 January 2023).
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: A new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Quiroga, R.; McConnell, S.; Johnson, M.C.; Casadevall, A. Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge. Int. J. Mol. Sci. 2023, 24, 2264. [Google Scholar] [CrossRef] [PubMed]
- Wolter, N.; Jassat, W.; Walaza, S.; Welch, R.; Moultrie, H.; Groome, M.; Amoako, D.G.; Everatt, J.; Bhiman, J.N.; Scheepers, C.; et al. Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: A data linkage study. Lancet 2022, 399, 437–446. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographics | Years | Total (n = 2547) | ||
|---|---|---|---|---|
| 2020 (n = 278) | 2021 (n = 1399) | 2022 (n = 870) | ||
| Age Groups (n (%)) | ||||
| <5 | 4 (1.4%) | 12 (0.9%) | 15 (1.7%) | 31 (1.2%) |
| 5–14 | 6 (2.2%) | 129 (9.2%) | 54 (6.2%) | 189 (7.7.%) |
| 15–24 | 26 (9.4%) | 234 (16.7%) | 119 (13.7%) | 379 (15.4%) |
| 25–44 | 141 (50.7%) | 611 (43.7%) | 410 (47.1%) | 1162 (47.3%) |
| 45–60 | 59 (21.2%) | 296 (21.2%) | 162 (18.6%) | 517 (21.0%) |
| >60 | 33 (3.2%) | 97 (6.9%) | 68 (7.8%) | 198 (8.1%) |
| Unknown * | 9 (3.2%) | 20 (1.4%) | 42 (4.8%) | 71 (2.9%) |
| Sex (n (%)) | ||||
| Female | 169 (60.8%) | 813 (58.1%) | 548 (63.0%) | 1531 (60.1%) |
| Male | 106 (38.1%) | 550 (39.3%) | 299 (34.4%) | 956 (37.5%) |
| Unknown * | 3 (1.1%) | 36 (2.6%) | 23 (2.6%) | 62 (2.4%) |
| Province (n (%)) | ||||
| Eastern Cape | 5 (1.8%) | 54 (3.7%) | 0 (0.0%) | 59 (2.3.%) |
| Gauteng | 264 (95.0%) | 1337 (95.6%) | 870 (100.0%) | 2471 (97.0%) |
| KwaZulu-Natal | 7 (2.5%) | 1 (0.1%) | 0 (0.0%) | 8 (0.3%) |
| Limpopo | 0 (0.0%) | 1 (0.1%) | 0 (0.0%) | 1 (0.1%) |
| Western Cape | 0 (0.0%) | 6 (0.4%) | 0 (0.0%) | 6 (0.4%) |
| Unknown * | 2 (0.7%) | 0 (0.0%) | 0 (0.0%) | 2 (0.1%) |
| Patient Status (n (%)) | ||||
| Community screening and test | 142 (51.5%) | 1150 (82.2%) | 536 (61.6%) | 1828 (71.8%) |
| Deceased | 0 (0.0%) | 4 (0.3%) | 11 (1.3%) | 15 (0.6%) |
| In-patient | 40 (14.4.%) | 77 (5.5%) | 92 (10.6%) | 209 (8.2%) |
| Out-patient | 93 (33.5%) | 168 (12.0%) | 231 (26.6%) | 492 (19.3%) |
| Unknown * | 3/278 (1.1%) | 0 (0.0%) | 0 (0.0%) | 3/2459 (0.1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramoney, K.; Mtileni, N.; Giandhari, J.; Naidoo, Y.; Ramphal, Y.; Pillay, S.; Ramphal, U.; Maharaj, A.; Tshiabuila, D.; Tegally, H.; et al. Molecular Epidemiology of SARS-CoV-2 during Five COVID-19 Waves and the Significance of Low-Frequency Lineages. Viruses 2023, 15, 1194. https://doi.org/10.3390/v15051194
Subramoney K, Mtileni N, Giandhari J, Naidoo Y, Ramphal Y, Pillay S, Ramphal U, Maharaj A, Tshiabuila D, Tegally H, et al. Molecular Epidemiology of SARS-CoV-2 during Five COVID-19 Waves and the Significance of Low-Frequency Lineages. Viruses. 2023; 15(5):1194. https://doi.org/10.3390/v15051194
Chicago/Turabian StyleSubramoney, Kathleen, Nkhensani Mtileni, Jennifer Giandhari, Yeshnee Naidoo, Yajna Ramphal, Sureshnee Pillay, Upasana Ramphal, Akhil Maharaj, Derek Tshiabuila, Houriiyah Tegally, and et al. 2023. "Molecular Epidemiology of SARS-CoV-2 during Five COVID-19 Waves and the Significance of Low-Frequency Lineages" Viruses 15, no. 5: 1194. https://doi.org/10.3390/v15051194