

m6A Regulates the Stability of Cellular Transcripts Required for Efficient KSHV Lytic Replication
 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mammalian Cell Culture
2.2. Antibodies and Plasmids
2.3. RNA Extraction, cDNA Synthesis and qPCR
2.4. Immunoblotting
2.5. Immunofluorescence
2.6. Luciferase Assays
2.7. Viral Reinfection Assays
2.8. Lentiviral Transduction
2.9. m6A Immunoprecipitation
2.10. RNA Stability Assays
2.11. Immunoprecipitation Assays
2.12. Quantitative Proteomics
2.13. Statistical Analysis
2.14. Data Availability
3. Results
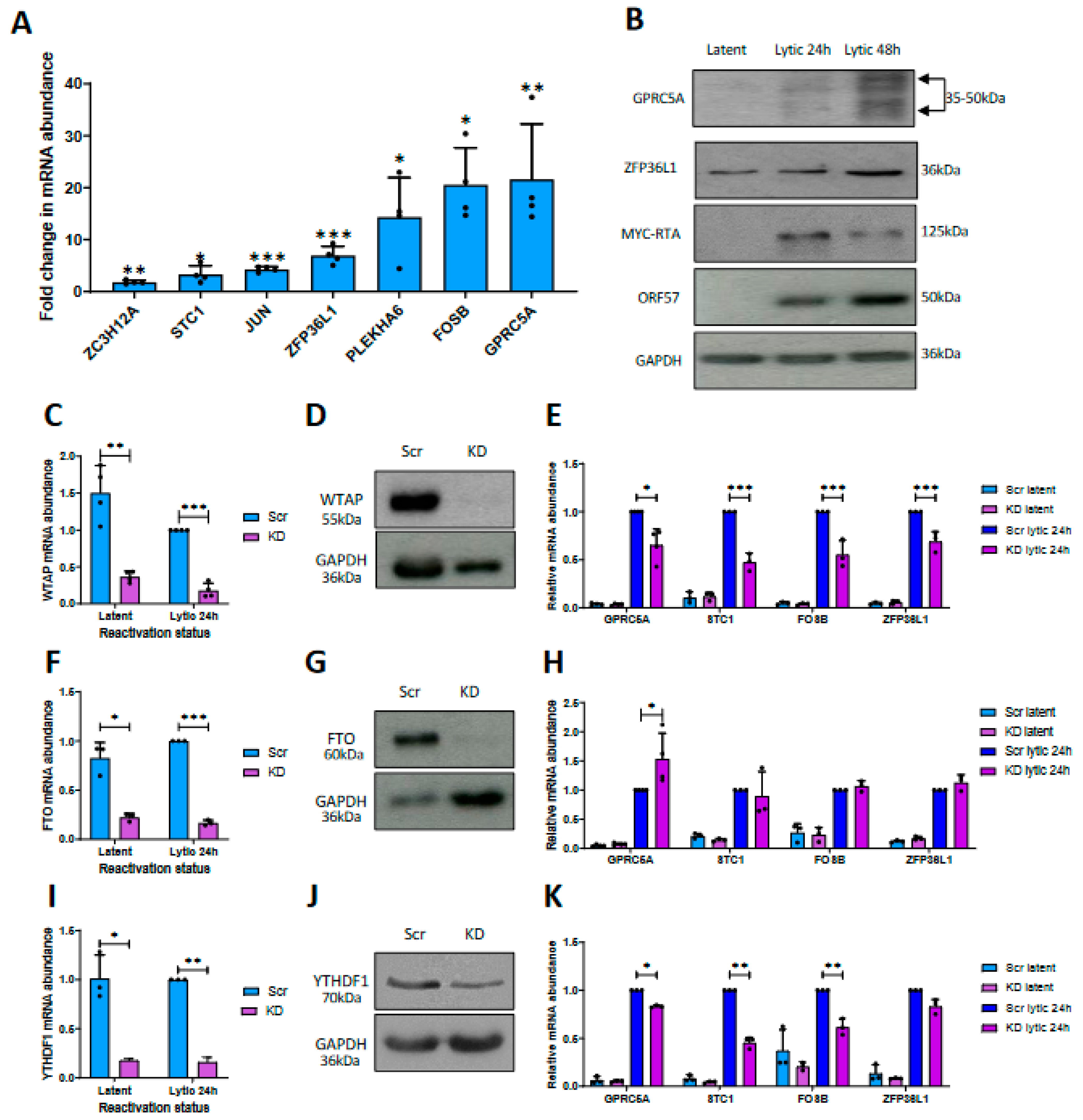
3.1. Differential m6A Status of Host Cell Transcripts Corresponds to Expression Levels during KSHV Lytic Replication
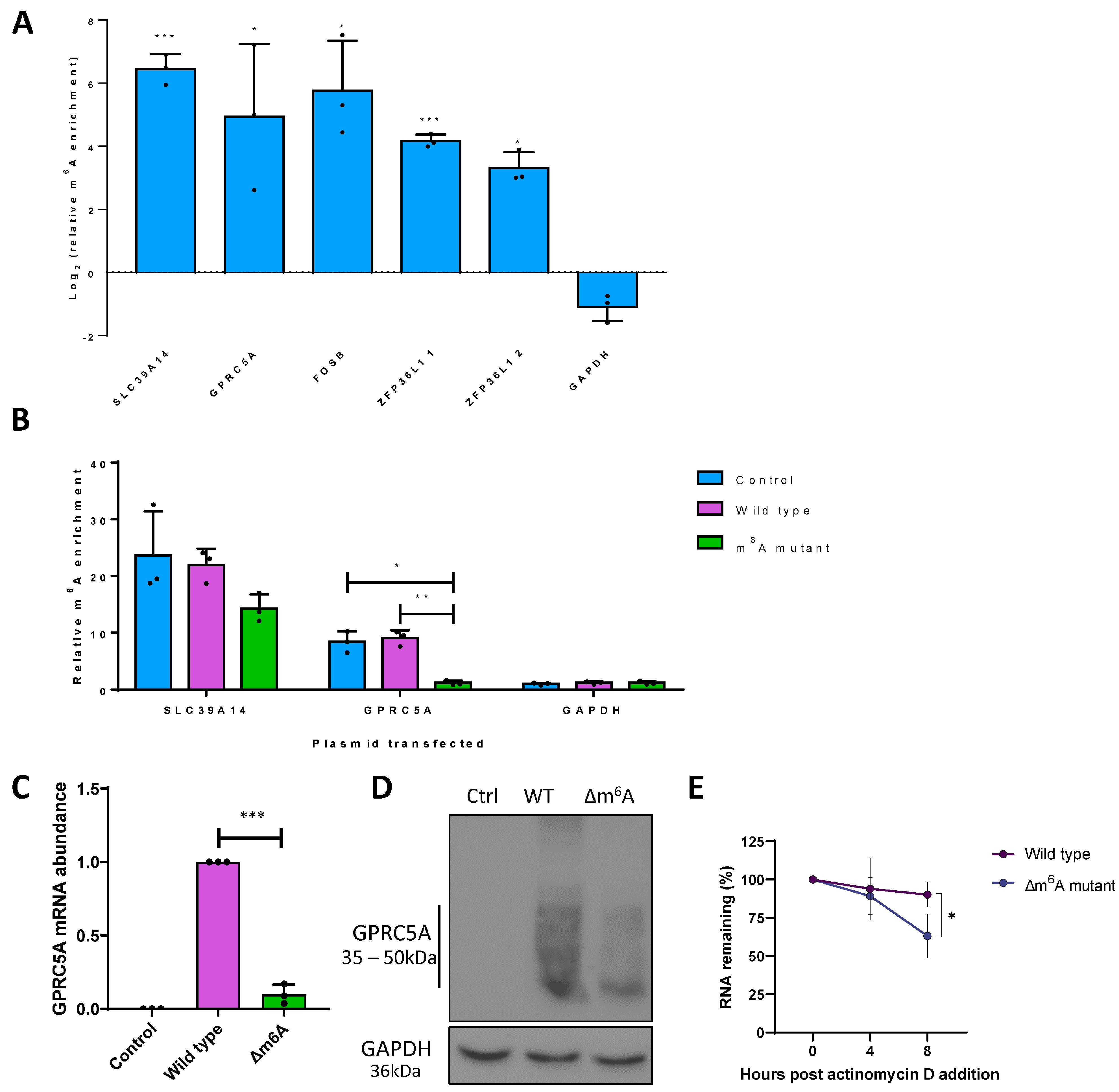
3.2. m6A Sites within GPRC5A mRNA Regulate Its Stability
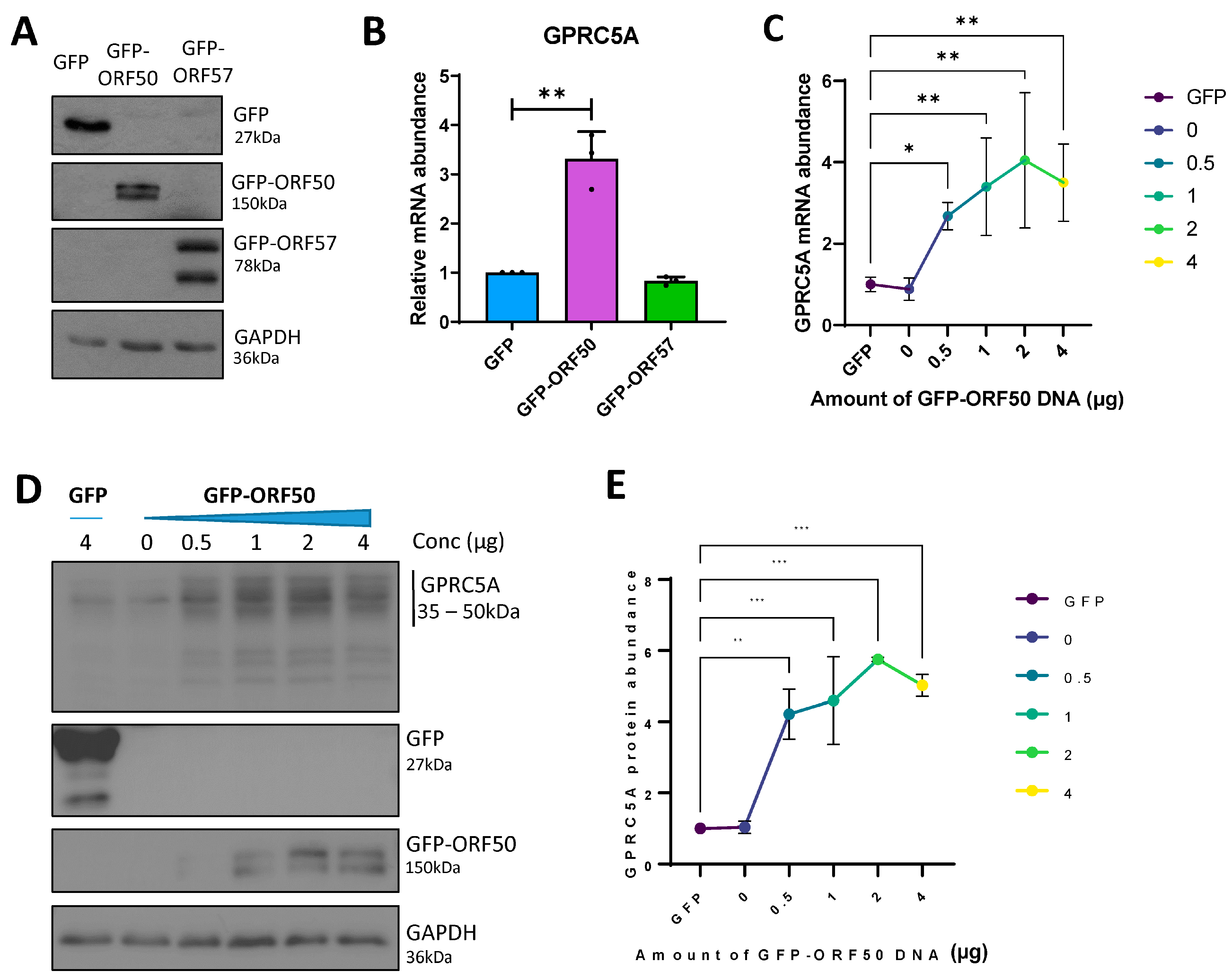
3.3. RTA Transactivates the GPRC5A Promoter
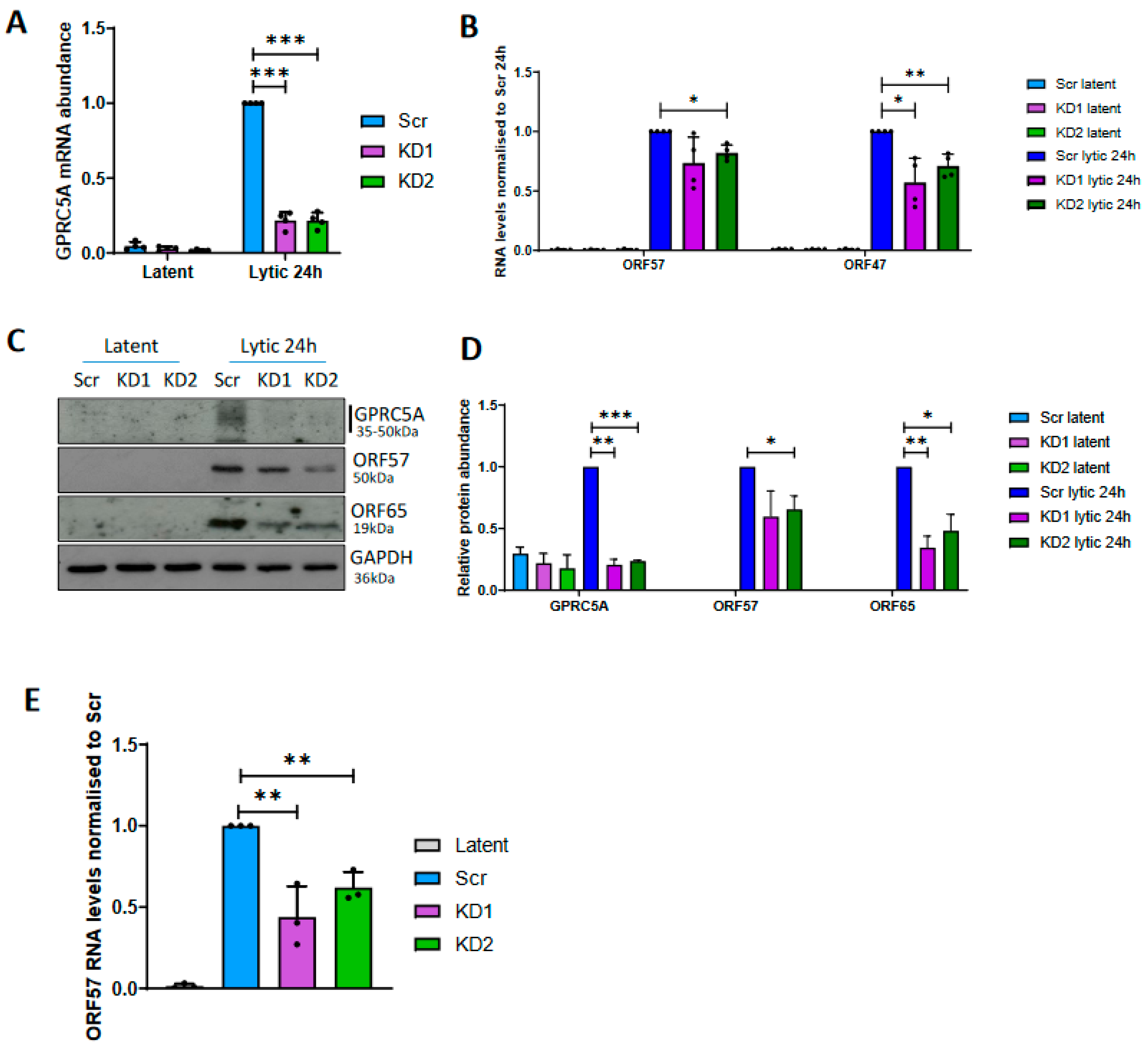
3.4. GPRC5A Is Required for Efficient KSHV Lytic Reactivation
3.5. GPRC5A Inhibits NFκB Signalling to Support KSHV Lytic Replication
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ganem, D. KSHV and the pathogenesis of Kaposi sarcoma: Listening to human biology and medicine. J. Clin. Investig. 2010, 120, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P.; Damania, B. Kaposi’s Sarcoma-Associated Herpesvirus (KSHV)-Associated Disease in the AIDS Patient: An Update. Cancer Treat. Res. 2019, 177, 63–80. [Google Scholar] [PubMed]
- Lange, P.; Damania, B. Kaposi Sarcoma-Associated Herpesvirus (KSHV). Trends Microbiol. 2020, 28, 236–237. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Cheng, F.; da Silva, S.R.; Tan, B.; Sorel, O.; Gruffaz, M.; Li, T.; Gao, S.J. Molecular Biology of KSHV in Relation to HIV/AIDS-Associated Oncogenesis. Cancer Treat. Res. 2019, 177, 23–62. [Google Scholar]
- Dittmer, D.; Lagunoff, M.; Renne, R.; Staskus, K.; Haase, A.; Ganem, D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1998, 72, 8309–8315. [Google Scholar] [CrossRef] [Green Version]
- Staskus, K.A.; Zhong, W.; Gebhard, K.; Herndier, B.; Wang, H.; Renne, R.; Beneke, J.; Pudney, J.; Anderson, D.J.; Ganem, D.; et al. Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 1997, 71, 715–719. [Google Scholar] [CrossRef] [Green Version]
- McClure, L.V.; Sullivan, C.S. Kaposi’s sarcoma herpes virus taps into a host microRNA regulatory network. Cell Host Microbe 2008, 3, 715–719. [Google Scholar] [CrossRef] [Green Version]
- Broussard, G.; Damania, B. Regulation of KSHV Latency and Lytic Reactivation. Viruses 2020, 12, 1034. [Google Scholar] [CrossRef]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef]
- Lukac, D.M.; Yuan, Y. Reactivation and lytic replication of KSHV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; National Library of Medicine: Bethesda, MD, USA, 2007. [Google Scholar]
- Purushothaman, P.; Dabral, P.; Gupta, N.; Sarkar, R.; Verma, S.C. KSHV Genome Replication and Maintenance. Front. Microbiol. 2016, 7, 54. [Google Scholar] [CrossRef] [Green Version]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giffin, L.; Damania, B. KSHV: Pathways to tumorigenesis and persistent infection. Adv. Virus Res. 2014, 88, 111–159. [Google Scholar] [PubMed] [Green Version]
- Nicholas, J. Human herpesvirus 8-encoded proteins with potential roles in virus-associated neoplasia. Front. Biosci. 2007, 12, 265–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manners, O.; Murphy, J.C.; Coleman, A.; Hughes, D.J.; Whitehouse, A. Contribution of the KSHV and EBV lytic cycles to tumourigenesis. Curr. Opin. Virol. 2018, 32, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Baquero-Perez, B.; Geers, D.; Diez, J. From A to m(6)A: The Emerging Viral Epitranscriptome. Viruses 2021, 13, 1049. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Salmon-Divon, M.; Amariglio, N.; Rechavi, G. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 2013, 8, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Meyer, K.D.; Jaffrey, S.R. Rethinking m(6)A Readers, Writers, and Erasers. Annu. Rev. Cell Dev. Biol. 2017, 33, 319–342. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m(6)A(m) in the 5’ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Theler, D.; Dominguez, C.; Blatter, M.; Boudet, J.; Allain, F.H. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: A reader of methylated RNA. Nucleic Acids Res. 2014, 42, 13911–13919. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [Green Version]
- Brocard, M.; Ruggieri, A.; Locker, N. m6A RNA methylation, a new hallmark in virus-host interactions. J. Gen. Virol. 2017, 98, 2207–2214. [Google Scholar] [CrossRef]
- Lichinchi, G.; Gao, S.; Saletore, Y.; Gonzalez, G.M.; Bansal, V.; Wang, Y.; Mason, C.E.; Rana, T.M. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 2016, 1, 16011. [Google Scholar] [CrossRef]
- Baquero-Perez, B.; Antanaviciute, A.; Yonchev, I.D.; Carr, I.M.; Wilson, S.A.; Whitehouse, A. The Tudor SND1 protein is an m(6)A RNA reader essential for replication of Kaposi’s sarcoma-associated herpesvirus. Elife 2019, 8, 47261. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.S.; McIntyre, A.B.R.; McFadden, M.J.; Roder, A.E.; Kennedy, E.M.; Gandara, J.A.; Hopcraft, S.E.; Quicke, K.M.; Vazquez, C.; Willer, J.; et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manners, O.; Baquero-Perez, B.; Whitehouse, A. m(6)A: Widespread regulatory control in virus replication. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Lin, X.; Gao, L.; Wei, Z.; Pei, Y.; Robertson, E.S. Attenuation of IFN signaling due to m(6)A modification of the host epitranscriptome promotes EBV lytic reactivation. J. Biomed. Sci. 2023, 30, 18. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, Y.; Watanabe, T.; Hara, Y.; Sato, Y.; Kimura, H.; Murata, T. EBV Exploits RNA m(6)A Modification to Promote Cell Survival and Progeny Virus Production During Lytic Cycle. Front. Microbiol. 2022, 13, 870816. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Chen, E.R.; Nilsen, T.W. Kaposi’s Sarcoma-Associated Herpesvirus Utilizes and Manipulates RNA N(6)-Adenosine Methylation To Promote Lytic Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.; Liu, H.; Zhang, S.; da Silva, S.R.; Zhang, L.; Meng, J.; Cui, X.; Yuan, H.; Sorel, O.; Zhang, S.W.; et al. Viral and cellular N(6)-methyladenosine and N(6),2’-O-dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat. Microbiol. 2018, 3, 108–120. [Google Scholar] [CrossRef]
- Hesser, C.R.; Karijolich, J.; Dominissini, D.; He, C.; Glaunsinger, B.A. N6-methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog. 2018, 14, e1006995. [Google Scholar] [CrossRef] [Green Version]
- Nwogu, N.; Boyne, J.R.; Dobson, S.J.; Poterlowicz, K.; Blair, G.E.; Macdonald, A.; Mankouri, J.; Whitehouse, A. Cellular sheddases are induced by Merkel cell polyomavirus small tumour antigen to mediate cell dissociation and invasiveness. PLoS Pathog. 2018, 14, e1007276. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Lu, M.; Gwack, Y.; Souvlis, J.; Zeichner, S.L.; Jung, J.U. Global changes in Kaposi’s sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 2003, 77, 4205–4220. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, D.J.; Hall, K.T.; Giles, M.S.; Calderwood, M.A.; Markham, A.F.; Whitehouse, A. The carboxy terminus of the herpesvirus saimiri ORF 57 gene contains domains that are required for transactivation and transrepression. J. Gen. Virol. 2000, 81, 2253–2658. [Google Scholar] [CrossRef]
- Gould, F.; Harrison, S.M.; Hewitt, E.W.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 2009, 83, 6727–6738. [Google Scholar] [CrossRef] [Green Version]
- Schumann, S.; Jackson, B.R.; Yule, I.; Whitehead, S.K.; Revill, C.; Foster, R.; Whitehouse, A. Targeting the ATP-dependent formation of herpesvirus ribonucleoprotein particle assembly as an antiviral approach. Nat. Microbiol. 2016, 2, 16201. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1-NFkappaB-IL-6 signalling axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef] [Green Version]
- Baquero-Perez, B.; Whitehouse, A. Hsp70 Isoforms Are Essential for the Formation of Kaposi’s Sarcoma-Associated Herpesvirus Replication and Transcription Compartments. PLoS Pathog. 2015, 11, e1005274. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, M.A.; Hall, K.T.; Matthews, D.A.; Whitehouse, A. The herpesvirus saimiri ORF73 gene product interacts with host-cell mitotic chromosomes and self-associates via its C terminus. J. Gen. Virol. 2004, 85, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Harper, K.L.; Mottram, T.J.; Anene, C.A.; Foster, B.; Patterson, M.R.; McDonnell, E.; Macdonald, A.; Westhead, D.; Whitehouse, A. Dysregulation of the miR-30c/DLL4 axis by circHIPK3 is essential for KSHV lytic replication. EMBO Rep. 2022, e54117. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.T.; Stevenson, A.J.; Goodwin, D.J.; Gibson, P.C.; Markham, A.F.; Whitehouse, A. The activation domain of herpesvirus saimiri R protein interacts with the TATA-binding protein. J. Virol. 1999, 73, 9756–9763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.C.; Harrington, E.M.; Schumann, S.; Vasconcelos, E.J.R.; Mottram, T.J.; Harper, K.L.; Aspden, J.L.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus induces specialised ribosomes to efficiently translate viral lytic mRNAs. Nat. Commun. 2023, 14, 300. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef]
- Glaunsinger, B.; Ganem, D. Highly selective escape from KSHV-mediated host mRNA shutoff and its implications for viral pathogenesis. J. Exp. Med. 2004, 200, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.; O’Hearn, P.M. Use of the red fluorescent protein as a marker of Kaposi’s sarcoma-associated herpesvirus lytic gene expression. Virology 2004, 325, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasman, L.; Krupalnik, V.; Viukov, S.; Mor, N.; Aguilera-Castrejon, A.; Schneir, D.; Bayerl, J.; Mizrahi, O.; Peles, S.; Tawil, S.; et al. Context-dependent functional compensation between Ythdf m(6)A reader proteins. Genes Dev. 2020, 34, 1373–1391. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Sepich-Poore, C.; Zhou, X.; Wei, J.; He, C. The mechanism underlying redundant functions of the YTHDF proteins. Genome Biol. 2023, 24, 17. [Google Scholar] [CrossRef]
- Guito, J.; Lukac, D.M. KSHV Rta Promoter Specification and Viral Reactivation. Front. Microbiol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, B.R.; Noerenberg, M.; Whitehouse, A. The Kaposi’s Sarcoma-Associated Herpesvirus ORF57 Protein and Its Multiple Roles in mRNA Biogenesis. Front. Microbiol. 2012, 3, 59. [Google Scholar] [CrossRef] [Green Version]
- Papp, B.; Motlagh, N.; Smindak, R.J.; Jin Jang, S.; Sharma, A.; Alonso, J.D.; Toth, Z. Genome-Wide Identification of Direct RTA Targets Reveals Key Host Factors for Kaposi’s Sarcoma-Associated Herpesvirus Lytic Reactivation. J. Virol. 2019, 93, e01978-18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Feng, H.; Xu, S.; Feng, P. Hijacking GPCRs by viral pathogens and tumor. Biochem. Pharmacol. 2016, 114, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Gao, Y.; Huang, Q.; Wang, Y.; Mo, X.; Wang, P.; Zhang, Y.; Xie, C.; Li, D.; Yao, J. Flotillin-1 Interacts With and Sustains the Surface Levels of TRPV2 Channel. Front. Cell Dev. Biol. 2021, 9, 634160. [Google Scholar] [CrossRef]
- Otto, G.P.; Nichols, B.J. The roles of flotillin microdomains-endocytosis and beyond. J. Cell Sci. 2011, 124 Pt 23, 3933–3940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, D.J.; Wood, J.J.; Jackson, B.R.; Baquero-Perez, B.; Whitehouse, A. NEDDylation is essential for Kaposi’s sarcoma-associated herpesvirus latency and lytic reactivation and represents a novel anti-KSHV target. PLoS Pathog. 2015, 11, e1004771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossmann, C.; Ganem, D. Effects of NFkappaB activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology 2008, 375, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.Z.; Chai, R.C.; Pang, B.; Chang, X.; An, S.Y.; Zhang, K.N.; Jiang, T.; Wang, Y.Z. METTL3 enhances the stability of MALAT1 with the assistance of HuR via m6A modification and activates NF-kappaB to promote the malignant progression of IDH-wildtype glioma. Cancer Lett. 2021, 511, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Villar, V.A.; Cuevas, S.; Zheng, X.; Jose, P.A. Localization and signaling of GPCRs in lipid rafts. Methods Cell Biol. 2016, 132, 3–23. [Google Scholar] [PubMed]
- Yang, L.; Ma, T.; Zhang, J. GPRC5A exerts its tumor-suppressive effects in breast cancer cells by inhibiting EGFR and its downstream pathway. Oncol. Rep. 2016, 36, 2983–2990. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Rigoutsos, I. The emerging roles of GPRC5A in diseases. Oncoscience 2014, 1, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ye, D.; Wang, T.; Guo, W.; Song, H.; Liao, Y.; Xu, D.; Zhu, H.; Zhang, Z.; Deng, J. Repression of GPRC5A is associated with activated STAT3, which contributes to tumor progression of head and neck squamous cell carcinoma. Cancer Cell Int. 2017, 17, 34. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Zhong, S.; Ye, X.; Liao, Y.; Yao, F.; Yang, X.; Sun, B.; Zhang, J.; Li, Q.; Gao, Y.; et al. EGFR phosphorylates and inhibits lung tumor suppressor GPRC5A in lung cancer. Mol. Cancer 2014, 13, 233. [Google Scholar] [CrossRef] [Green Version]
- Brown, H.J.; Song, M.J.; Deng, H.; Wu, T.T.; Cheng, G.; Sun, R. NF-kappaB inhibits gammaherpesvirus lytic replication. J. Virol. 2003, 77, 8532–8540. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manners, O.; Baquero-Perez, B.; Mottram, T.J.; Yonchev, I.D.; Trevelyan, C.J.; Harper, K.L.; Menezes, S.; Patterson, M.R.; Macdonald, A.; Wilson, S.A.; et al. m6A Regulates the Stability of Cellular Transcripts Required for Efficient KSHV Lytic Replication. Viruses 2023, 15, 1381. https://doi.org/10.3390/v15061381
Manners O, Baquero-Perez B, Mottram TJ, Yonchev ID, Trevelyan CJ, Harper KL, Menezes S, Patterson MR, Macdonald A, Wilson SA, et al. m6A Regulates the Stability of Cellular Transcripts Required for Efficient KSHV Lytic Replication. Viruses. 2023; 15(6):1381. https://doi.org/10.3390/v15061381
Chicago/Turabian StyleManners, Oliver, Belinda Baquero-Perez, Timothy J. Mottram, Ivaylo D. Yonchev, Christopher J. Trevelyan, Katherine L. Harper, Sarah Menezes, Molly R. Patterson, Andrew Macdonald, Stuart A. Wilson, and et al. 2023. "m6A Regulates the Stability of Cellular Transcripts Required for Efficient KSHV Lytic Replication" Viruses 15, no. 6: 1381. https://doi.org/10.3390/v15061381