
High Genetic Diversity of HIV-1 and Active Transmission Clusters among Male-to-Male Sexual Contacts (MMSCs) in Zhuhai, China
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Population
2.2. PCR Amplification and Recency Testing
2.3. HIV Sequencing and Subtyping
2.4. Phylogenetic and Clustering Analysis
2.5. Statistical Analysis
3. Results
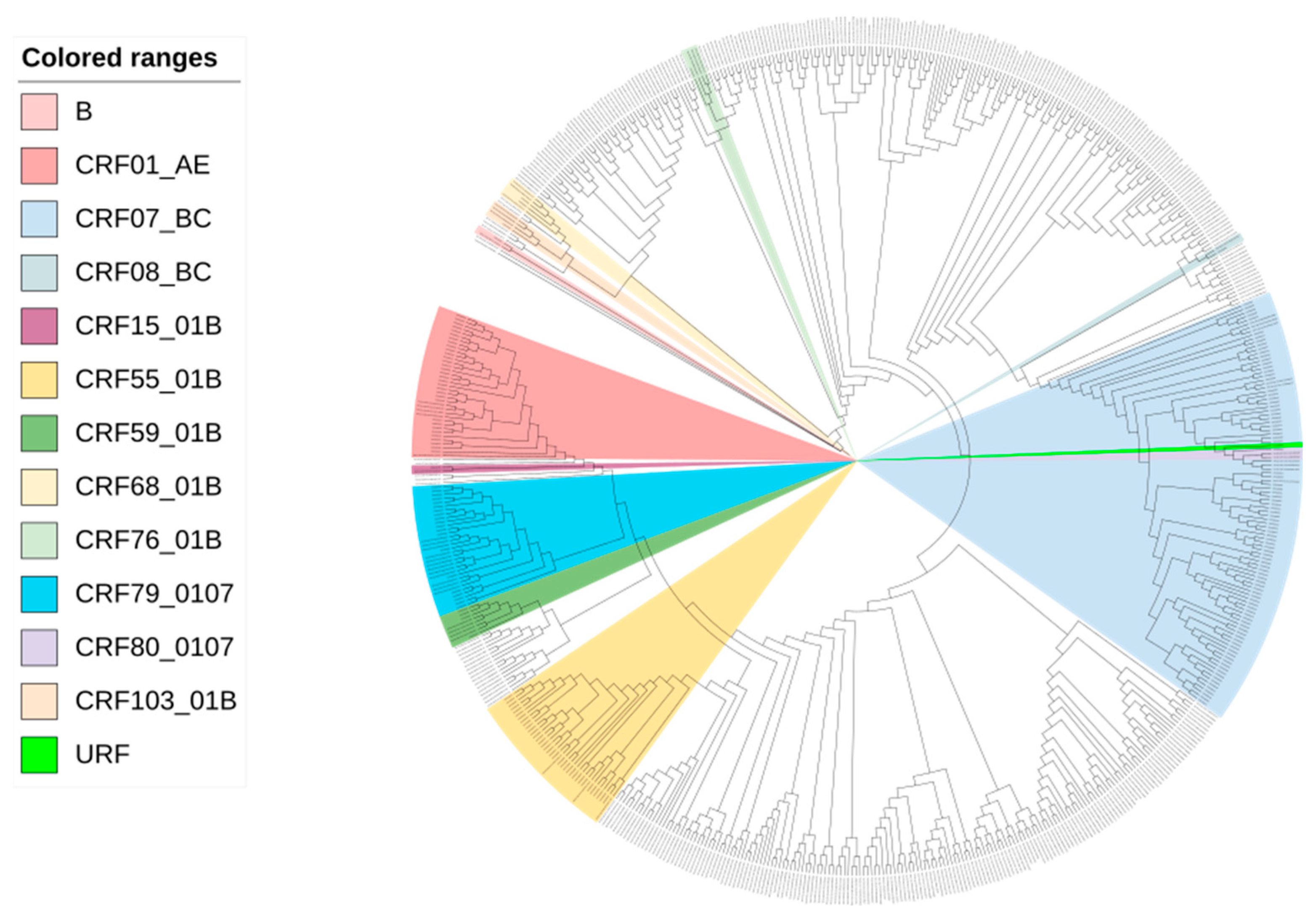
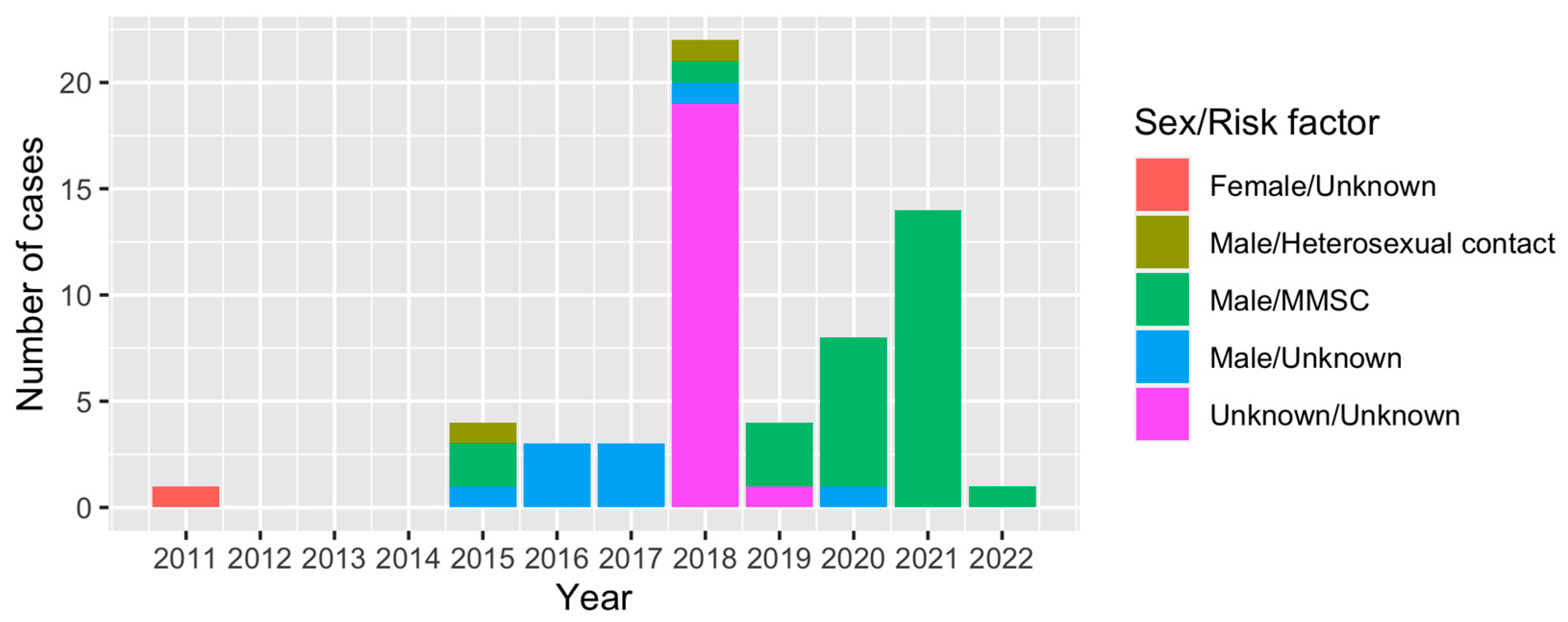
3.1. Demographics and HIV Genotyping of Study Subjects
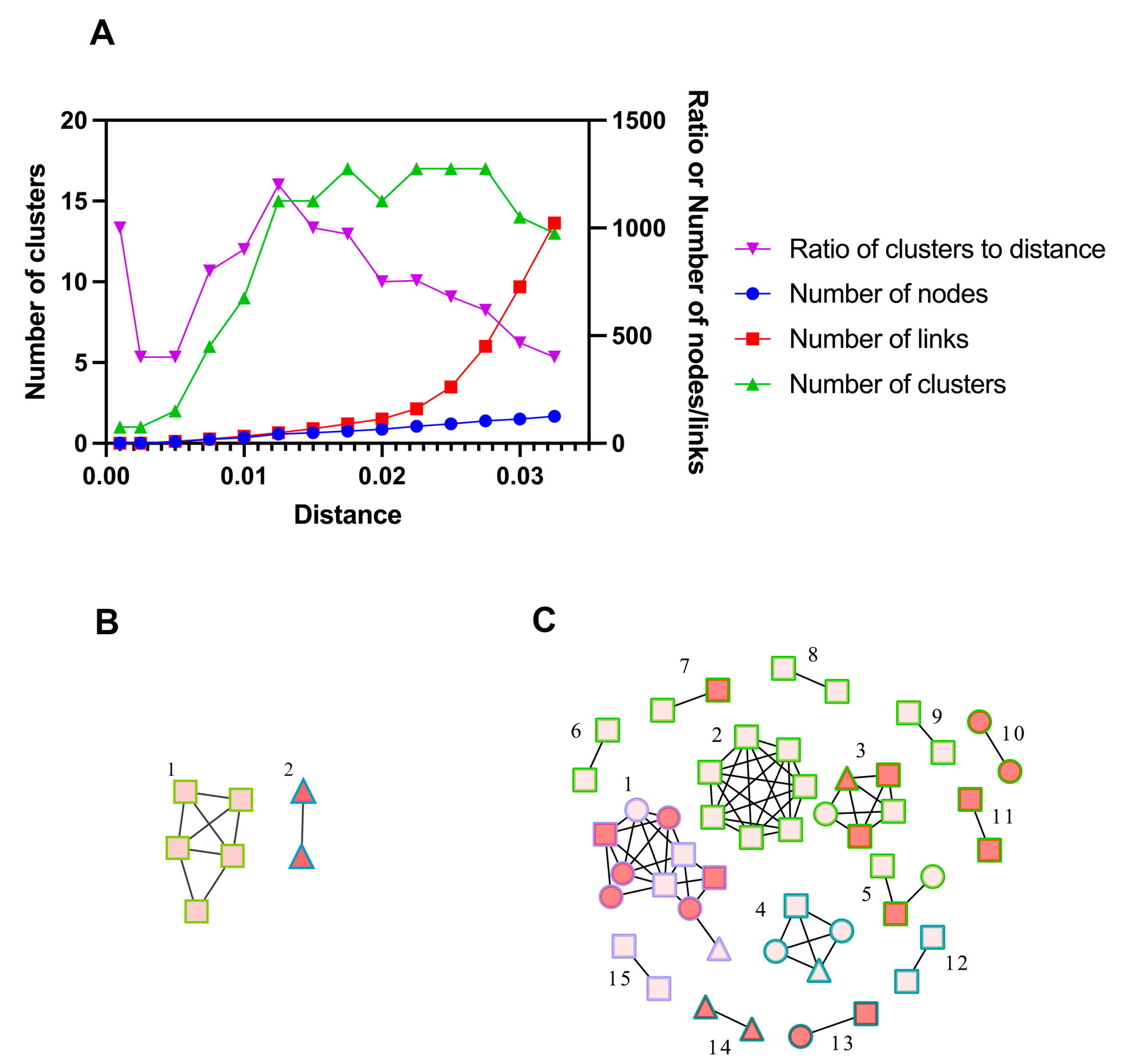
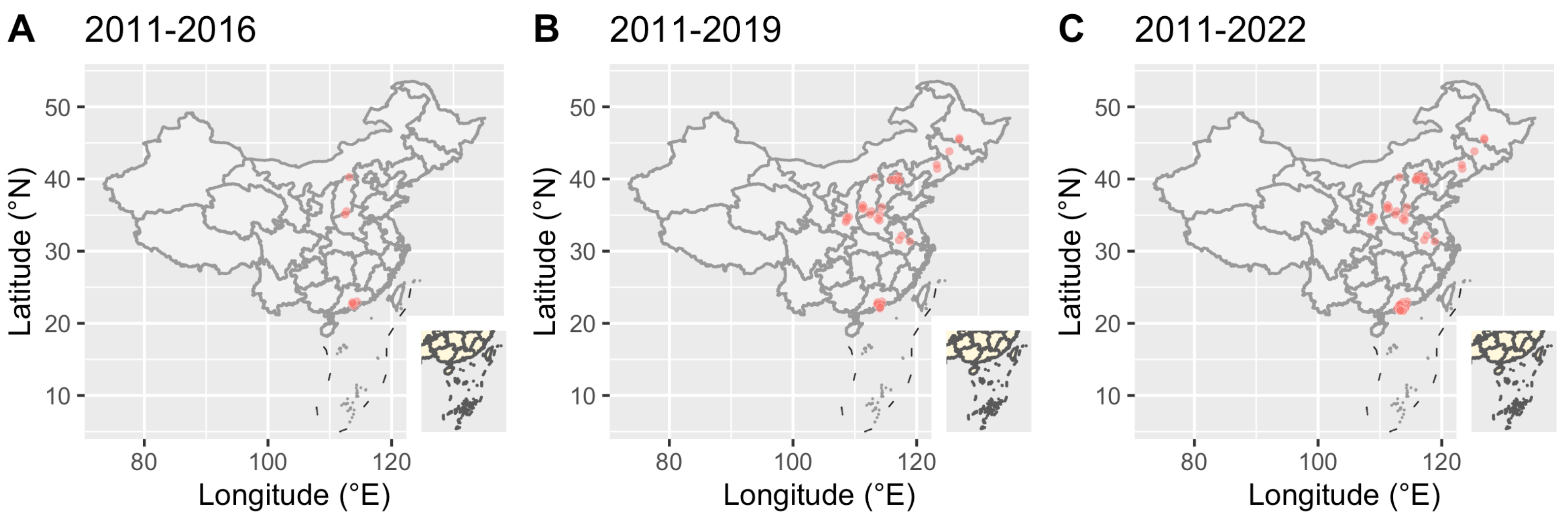
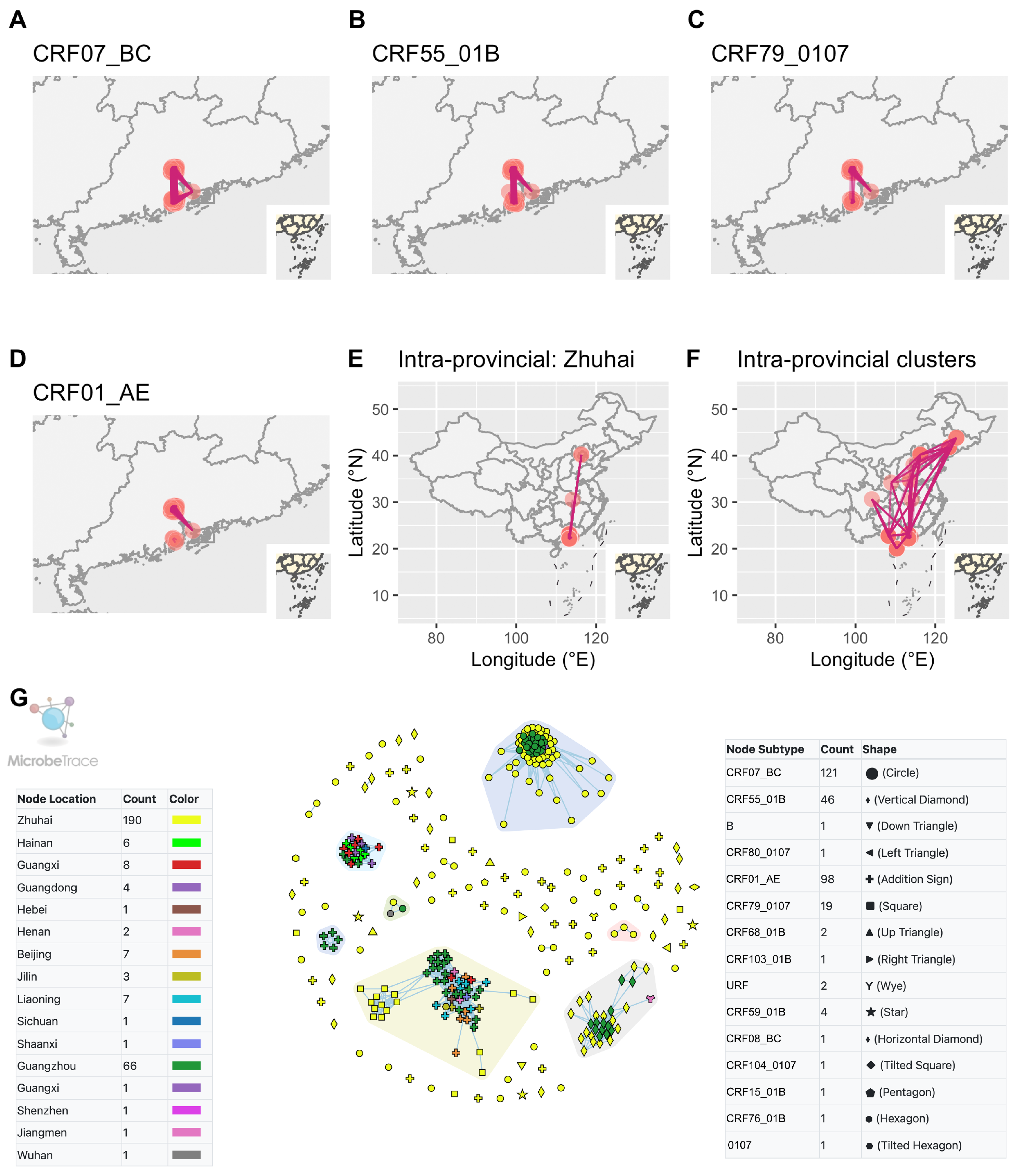
3.2. Molecular Network Analysis and Active Transmission Clusters
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS. Global HIV & AIDS Statistics—Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 3 August 2023).
- WHO. Consolidated Guidelines on HIV Prevention, Diagnosis, Treatment and Care for Key Populations. Available online: https://www.who.int/publications/i/item/9789241511124 (accessed on 20 May 2022).
- Dong, M.J.; Peng, B.; Liu, Z.F.; Ye, Q.N.; Liu, H.; Lu, X.L.; Zhang, B.; Chen, J.J. The prevalence of HIV among MSM in China: A large-scale systematic analysis. BMC Infect. Dis. 2019, 19, 1000. [Google Scholar] [CrossRef]
- National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention. Detecting and Responding to HIV Transmission Clusters a Guide For Health Departments. Available online: https://www.cdc.gov/hiv/pdf/funding/announcements/ps18-1802/CDC-HIV-PS18-1802-AttachmentE-Detecting-Investigating-and-Responding-to-HIV-Transmission-Clusters.pdf (accessed on 20 May 2022).
- Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: Pathogenetic and therapeutic implications. J. Virol. 2007, 81, 10209–10219. [Google Scholar] [CrossRef]
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV subtype diversity worldwide. Curr. Opin. HIV AIDS 2019, 14, 153–160. [Google Scholar] [CrossRef]
- Delwart, E.L.; Shpaer, E.G.; Louwagie, J.; McCutchan, F.E.; Grez, M.; Rübsamen-Waigmann, H.; Mullins, J.I. Genetic relationships determined by a DNA heteroduplex mobility assay: Analysis of HIV-1 env genes. Science 1993, 262, 1257–1261. [Google Scholar] [CrossRef]
- Li, X.; Li, W.; Zhong, P.; Fang, K.; Zhu, K.; Musa, T.H.; Song, Y.; Du, G.; Gao, R.; Guo, Y.; et al. Nationwide Trends in Molecular Epidemiology of HIV-1 in China. AIDS Res. Hum. Retroviruses 2016, 32, 851–859. [Google Scholar] [CrossRef]
- Xuan, Q.; Liang, S.; Qin, W.; Yang, S.; Zhang, A.M.; Zhao, T.; Su, H.; Xia, Z.; Wang, B.; Xia, X. High prevalence of HIV-1 transmitted drug resistance among therapy-naïve Burmese entering travelers at Dehong ports in Yunnan, China. BMC Infect. Dis. 2018, 18, 211. [Google Scholar] [CrossRef]
- Sun, L.; Jia, L.; Liu, Y.; Han, J.; Li, J.; Li, H.; Li, L. Multiple HIV-1 Subtypes Were Found Circulating in Shijingshan District of Beijing, China. AIDS Res. Hum. Retroviruses 2019, 35, 494–499. [Google Scholar] [CrossRef]
- Stephenson, K.E.; Wagh, K.; Korber, B.; Barouch, D.H. Vaccines and Broadly Neutralizing Antibodies for HIV-1 Prevention. Annu. Rev. Immunol. 2020, 38, 673–703. [Google Scholar] [CrossRef]
- Zhou, L.; Wei, Q.; Huang, H.; Li, X. Genotypes and drug resistance of HIV-1 in Zhuhai, 2018. Pract. Prev. Med. 2020, 12, 1409–1411. [Google Scholar]
- Murphy, G.; Parry, J.V. Assays for the detection of recent infections with human immunodeficiency virus type 1. Wkly. Releases (1997–2007) 2008, 13, 18966. [Google Scholar] [CrossRef]
- UNAIDS/WHO. When and How to Use Assays for Recent Infection to Estimate HIV Incidence at a Population Level. Available online: https://apps.who.int/iris/bitstream/handle/10665/44612/9789241501675_eng.pdf?msclkid=6e7ad557cf9111ec8d4ed8ccb3f8e001 (accessed on 9 May 2022).
- Hall, H.I.; Song, R.; Rhodes, P.; Prejean, J.; An, Q.; Lee, L.M.; Karon, J.; Brookmeyer, R.; Kaplan, E.H.; McKenna, M.T.; et al. Estimation of HIV incidence in the United States. JAMA 2008, 300, 520–529. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, M.; Ni, M.; Duan, S.; Wang, Y.; Feng, J.; Xiao, Y.; Dong, Y.; Wang, D.; Han, M.; et al. HIV-1 incidence estimates using IgG-capture BED-enzyme immunoassay from surveillance sites of injection drug users in three cities of China. Aids 2007, 21 (Suppl. 8), S47–S51. [Google Scholar] [CrossRef]
- Soodla, P.; Simmons, R.; Huik, K.; Pauskar, M.; Jõgeda, E.L.; Rajasaar, H.; Kallaste, E.; Maimets, M.; Avi, R.; Murphy, G.; et al. HIV incidence in the Estonian population in 2013 determined using the HIV-1 limiting antigen avidity assay. HIV Med. 2017, 19, 33–41. [Google Scholar] [CrossRef]
- Wu, D.; Zhou, Y.; Yang, N.; Huang, S.; He, X.; Tucker, J.; Li, X.; Smith, K.M.; Ritchwood, T.; Jiang, X.; et al. Social Media-Based Secondary Distribution of Human Immunodeficiency Virus/Syphilis Self-testing Among Chinese Men Who Have Sex with Men. Clin. Infect. Dis. 2020, 73, e2251–e2257. [Google Scholar] [CrossRef]
- Li, W.; Huang, S.; Yao, G.; Zhou, Y.; Dai, W.; Li, X.; Du, M. Epidemiological characteristics of HIV/AIDS among MSM in Zhuhai city, 2002–2018. J. Trop. Med. 2020, 20, 253–256. [Google Scholar]
- Tang, X.J.; Duan, L.J.; Liang, W.L.; Cheng, S.; Dong, T.L.; Xie, Z.; Liu, K.M.; Yu, F.; Chen, Z.H.; Mi, G.D.; et al. [Application of limiting antigen avidity enzyme immunoassay for estimating HIV-1 incidence in men who have sex with men]. Zhonghua Liuxingbingxue Zazhi 2022, 43, 72–77. [Google Scholar] [CrossRef]
- Otecko, N.; Inzaule, S.; Odhiambo, C.; Otieno, G.; Opollo, V.; Morwabe, A.; Were, K.; Ndiege, K.; Otieno, F.; Kim, A.A.; et al. Viral and Host Characteristics of Recent and Established HIV-1 Infections in Kisumu based on a Multiassay Approach. Sci. Rep. 2016, 6, 37964. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.C.; Halpern, A.L.; Macken, C.; Korber, B.T. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res. Hum. Retroviruses 1995, 11, 1413–1416. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef]
- Li, M.; Zhou, J.; Zhang, K.; Yuan, Y.; Zhao, J.; Cui, M.; Yin, D.; Wen, Z.; Chen, Z.; Li, L.; et al. Characteristics of genotype, drug resistance, and molecular transmission network among newly diagnosed HIV-1 infections in Shenzhen, China. J. Med. Virol. 2023, 95, e28973. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Boyles, A.; Shankar, A.; Kim, J.; Knyazev, S.; Cintron, R.; Switzer, W.M. MicrobeTrace: Retooling molecular epidemiology for rapid public health response. PLOS Comput. Biol. 2021, 17, e1009300. [Google Scholar] [CrossRef]
- Fischer, W.; Giorgi, E.E.; Chakraborty, S.; Nguyen, K.; Bhattacharya, T.; Theiler, J.; Goloboff, P.A.; Yoon, H.; Abfalterer, W.; Foley, B.T.; et al. HIV-1 and SARS-CoV-2: Patterns in the evolution of two pandemic pathogens. Cell Host Microbe 2021, 29, 1093–1110. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Yang, Y.; Liang, N.; Liang, H.; Chen, Y.; Lin, Z.; Chen, T.; Tan, W.; Yang, Y.; Huang, R.; et al. Transmission network and phylogenetic analysis reveal older male-centered transmission of CRF01_AE and CRF07_BC in Guangxi, China. Emerg. Microbes Infect. 2022, 12, 1–48. [Google Scholar] [CrossRef]
- Yuan, D.; Yu, B.; Liang, S.; Fei, T.; Tang, H.; Kang, R.; Li, Y.; Ye, L.; Jia, P.; Yang, S. HIV-1 genetic transmission networks among people living with HIV/AIDS in Sichuan, China: A genomic and spatial epidemiological analysis. Lancet Reg. Heal. West. Pac. 2022, 18, 100318. [Google Scholar] [CrossRef]
- Lan, Y.; He, X.; Li, L.; Zhou, P.; Huang, X.; Deng, X.; Li, J.; Fan, Q.; Li, F.; Tang, X.; et al. Complicated genotypes circulating among treatment naïve HIV-1 patients in Guangzhou, China. Infect. Genet. Evol. 2020, 87, 104673. [Google Scholar] [CrossRef]
- Zhang, D.; Zheng, C.; Li, H.; Li, H.; Liu, Y.; Wang, X.; Jia, L.; Chen, L.; Yang, Z.; Gan, Y.; et al. Molecular surveillance of HIV-1 newly diagnosed infections in Shenzhen, China from 2011 to 2018. J. Infect. 2021, 83, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, Y.; Li, F.; Xue, Z.; Hu, J.; Xing, H.; Ruan, Y.; Shao, Y.; Wu, Y.; Wang, H.; et al. Genome Sequence of a Novel HIV-1 Circulating Recombinant Form (CRF79_0107) Identified from Shanxi, China. AIDS Res. Hum. Retroviruses 2017, 33, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Gan, M.; Zheng, S.; Hao, J.; Ruan, Y.; Liao, L.; Shao, Y.; Feng, Y.; Xing, H. The prevalence of CRF55_01B among HIV-1 strain and its connection with traffic development in China. Emerg. Microbes Infect. 2021, 10, 256–265. [Google Scholar] [CrossRef]
- Lundgren, J.D.; Babiker, A.G.; Gordin, F.; Emery, S.; Grund, B.; Sharma, S.; Avihingsanon, A.; Cooper, D.A.; Fätkenheuer, G.; Llibre, J.M.; et al. Initiation of Antiretroviral Therapy in Early Asymptomatic HIV Infection. N. Engl. J. Med. 2015, 373, 795–807. [Google Scholar]
- Zhao, Y.; Han, M.; Ma, Y.; Li, D. Progress Towards the 90-90-90 Targets for Controlling HIV—China, 2018. China CDC Wkly. 2019, 1, 5–7. [Google Scholar] [CrossRef]
- Na, H. New progress of AIDS epidemiology in China. Chin. J. Dis. Control Prev. 2021, 25, 1365–1368+1480. [Google Scholar]
- Zheng, D.; Zhu, J. Antiretroviral Treatment Coverage of 96% among AIDS Patients in Zhuhai. Available online: http://ep.ycwb.com/epaper/ywdf/html/2021-12/02/content_792_450972.htm (accessed on 2 December 2021).
- Wertheim, J.O.; Oster, A.M.; Switzer, W.M.; Zhang, C.; Panneer, N.; Campbell, E.; Saduvala, N.; Johnson, J.A.; Heneine, W. Natural selection favoring more transmissible HIV detected in United States molecular transmission network. Nat. Commun. 2019, 10, 5788. [Google Scholar] [CrossRef]
- Little, S.J.; Chen, T.; Wang, R.; Anderson, C.; Pond, S.K.; Nakazawa, M.; Mathews, W.C.; DeGruttola, V.; Smith, D.M. Effective Human Immunodeficiency Virus Molecular Surveillance Requires Identification of Incident Cases of Infection. Clin. Infect. Dis. 2021, 73, 842–849. [Google Scholar] [CrossRef]
- Zhou, S.; Sizemore, S.; Moeser, M.; Zimmerman, S.; Samoff, E.; Mobley, V.; Frost, S.; Cressman, A.; Clark, M.; Skelly, T.; et al. Near Real-Time Identification of Recent Human Immunodeficiency Virus Transmissions, Transmitted Drug Resistance Mutations, and Transmission Networks by Multiplexed Primer ID-Next-Generation Sequencing in North Carolina. J. Infect. Dis. 2021, 223, 876–884. [Google Scholar] [CrossRef]
- Truong, H.M.; Pipkin, S.; O’Keefe, K.J.; Louie, B.; Liegler, T.; McFarland, W.; Grant, R.M.; Bernstein, K.; Scheer, S. Brief Report: Recent Infection, Sexually Transmitted Infections, and Transmission Clusters Frequently Observed Among Persons Newly Diagnosed With HIV in San Francisco. J. Acquir. Immune Defic. Syndr. 2015, 69, 606–609. [Google Scholar] [CrossRef]
- Hilldorfer, B.B.; Cillo, A.R.; Besson, G.J.; Bedison, M.A.; Mellors, J.W. New tools for quantifying HIV-1 reservoirs: Plasma RNA single copy assays and beyond. Curr. HIV/AIDS Rep. 2012, 9, 91–100. [Google Scholar] [CrossRef]
- Broyles, L.N.; Luo, R.; Boeras, D.; Vojnov, L. The risk of sexual transmission of HIV in individuals with low-level HIV viraemia: A systematic review. Lancet 2023, 402, 464–471. [Google Scholar] [CrossRef]
- Gonzalo-Gil, E.; Ikediobi, U.; Sutton, R.E. Mechanisms of Virologic Control and Clinical Characteristics of HIV+ Elite/Viremic Controllers. Yale J. Biol. Med. 2017, 90, 245–259. [Google Scholar] [PubMed]
- Gan Mengze, D.A.; Kang, R.; Liao, L.; Ruan, Y.; Shao, Y.; Feng, Y.; Xing, H. Characteristics of inter-provincial transmission of newly reported HIV-1 infected people in 2018. Chin. J. AIDS STD 2021, 27, 115–120. [Google Scholar]
- Longosz, A.F.; Mehta, S.H.; Kirk, G.D.; Margolick, J.B.; Brown, J.; Quinn, T.C.; Eshleman, S.H.; Laeyendecker, O. Incorrect identification of recent HIV infection in adults in the United States using a limiting-antigen avidity assay. AIDS 2014, 28, 1227–1232. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. What Can Increase the Risk of Getting or Transmitting HIV? Available online: https://www.cdc.gov/hiv/basics/hiv-transmission/increase-hiv-risk.html (accessed on 14 September 2023).
- Tanser, F.; Vandormael, A.; Cuadros, D.; Phillips, A.N.; de Oliveira, T.; Tomita, A.; Bärnighausen, T.; Pillay, D. Effect of population viral load on prospective HIV incidence in a hyperendemic rural African community. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Morgan, E.; Skaathun, B.; Nikolopoulos, G.K.; Paraskevis, D.; Williams, L.D.; Smyrnov, P.; Friedman, S.R.; Schneider, J.A. A Network Intervention to Locate Newly HIV Infected Persons Within MSM Networks in Chicago. AIDS Behav. 2018, 23, 15–20. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequences (5′-3′) | Position in HBX2 |
|---|---|---|
| HR26871 | TTAAAGCCAGGAATGGATGG | 2583–2602 |
| HR26872 | TCAGGATGGAGTTCATACCCC | 3259–3239 |
| HR26873 | TGGAAAGGATCACCAGCAATA | 3006–3026 |
| HR27372 | TGTTAACTGTCTTACATCATTAGTGTG | 3704–3681 |
| Variable | Level | Long-Term Infection (n = 153, 80.5%) | Recent Infection (n = 37, 19.5%) | Total (n = 190, 100%) | p |
|---|---|---|---|---|---|
| Age (year) | 18~35 | 96 (62.7) | 28 (75.7) | 124 (65.3) | 0.197 |
| >35 | 57 (37.3) | 9 (24.3) | 66 (34.7) | ||
| Ethnicity | Han | 141 (92.2) | 33 (89.2) | 174 (91.6) | 0.520 1 |
| Others | 12 (7.8) | 4 (10.8) | 16 (8.4) | ||
| Marriage | Single | 109 (71.2) | 29 (78.4) | 138 (72.6) | 0.193 1 |
| Married | 25 (16.3) | 2 (5.4) | 27 (14.2) | ||
| Separated or divorced | 19 (12.4) | 6 (16.2) | 25 (13.2) | ||
| Education | Junior high school and below | 42 (27.5) | 7 (18.9) | 49 (25.8) | 0.493 |
| High school | 46 (30.1) | 14 (37.8) | 60 (31.6) | ||
| College degree or above | 65 (42.5) | 16 (43.2) | 81 (42.6) | ||
| Occupation | Office worker | 61 (39.9) | 11 (29.7) | 72 (37.9) | 0.195 1 |
| Housework or unemployment | 30 (19.6) | 7 (18.9) | 37 (19.5) | ||
| Ordinary worker or farmer | 28 (18.3) | 6 (16.2) | 34 (17.9) | ||
| Sexual worker | 19 (12.4) | 4 (10.8) | 23 (12.1) | ||
| Student | 8 (5.2) | 7 (18.9) | 15 (7.9) | ||
| Others | 7 (4.6) | 2 (5.4) | 9 (4.7) | ||
| Subtype | CRF07_BC | 75 (49.0) | 17 (45.9) | 92 (48.4) | 0.041 1 |
| CRF55_01B | 26 (17.0) | 8 (21.6) | 34 (17.9) | ||
| CRF01_AE | 29 (19.0) | 2 (5.4) | 31 (16.3) | ||
| CRF79_0107 | 11 (7.2) | 8 (21.6) | 19 (10.0) | ||
| Others | 12 (7.8) | 2 (5.4) | 14 (7.4) | ||
| Cluster 2 | No | 123 (80.4) | 18 (48.6) | 141 (74.2) | <0.001 |
| Yes | 30 (19.6) | 19 (51.4) | 49 (25.8) |
| Viral Load (copies/mL) | Nonclustering 2 (n = 141, 74.2%) | Clustering 2 (n = 49, 25.8%) | Total (n = 190, 100%) | p 1 |
|---|---|---|---|---|
| <1000 | 52 (36.9) | 10 (20.4) | 62 (32.6) | 0.091 |
| 1000~10,000 | 10 (7.1) | 3 (6.1) | 13 (6.8) | |
| ≥10,000 | 72 (51.1) | 35 (71.4) | 107 (56.3) | |
| Missing | 7 (5.0) | 1 (2.0) | 8 (4.2) |
| Variable | Level | Univariable Analysis | Multivariable Analysis | ||
|---|---|---|---|---|---|
| OR (95%CI) | p | aOR (95%CI) | p | ||
| Age (year) | 18~35 | ref. | |||
| >35 | 0.54 [0.23, 1.19] | 0.142 | |||
| Ethnicity | Han | ||||
| Others | 1.42 [0.38, 4.39] | 0.561 | |||
| Marriage | Single | ref. | |||
| Married | 0.30 [0.05, 1.09] | 0.116 | |||
| Separated or divorced | 1.19 [0.40, 3.10] | 0.738 | |||
| Education | Junior high school and below | ref. | |||
| High school | 1.83 [0.69, 5.22] | 0.237 | |||
| College degree or above | 1.48 [0.58, 4.12] | 0.430 | |||
| Occupation | Office worker | ref. | ref. | ||
| Housework or unemployment | 1.29 [0.44, 3.63] | 0.628 | 1.71 [0.53, 5.41] | 0.359 | |
| Worker or farmer | 1.19 [0.38, 3.46] | 0.757 | 1.96 [0.57, 6.51] | 0.270 | |
| Sexual worker | 1.17 [0.30, 3.87] | 0.809 | 1.71 [0.40, 6.60] | 0.442 | |
| Student | 4.85 [1.44, 16.45] | 0.010 | 6.47 [1.71, 25.66] | 0.006 | |
| Others | 1.58 [0.22, 7.66] | 0.595 | 4.03 [0.51, 22.87] | 0.134 | |
| Subtype | CRF07_BC | ref. | ref. | ||
| CRF55_01B | 1.36 [0.50, 3.44] | 0.529 | 1.38 [0.46, 3.91] | 0.546 | |
| CRF01_AE | 0.30 [0.05, 1.15] | 0.127 | 0.47 [0.07, 1.97] | 0.355 | |
| CRF79_0107 | 3.21 [1.10, 9.20] | 0.030 | 3.03 [0.95, 9.56] | 0.057 | |
| Others | 0.74 [0.11, 3.04] | 0.704 | 1.29 [0.18, 5.95] | 0.764 | |
| Cluster * | No | ref. | ref. | ||
| Yes | 4.33 [2.03, 9.33] | <0.001 | 3.64 [1.51, 9.01] | 0.004 | |
| Variable | Level | MMSCs with High Linkage (n = 13, 26.5%) | MMSCs with Low Linkage (n = 36, 73.4%) | Total (n = 49, 100%) | p * |
|---|---|---|---|---|---|
| Age (year) | 18~35 | 13 (100.0) | 25 (69.4) | 38 (77.6) | 0.024 |
| >35 | 0 (0.0) | 11 (30.6) | 11 (22.4) | ||
| Ethnicity | Han | 9 (69.2) | 34 (94.4) | 43 (87.8) | 0.036 |
| Others | 4 (30.8) | 2 (5.6) | 6 (12.2) | ||
| Marriage | Single | 13 (100.0) | 23 (63.9) | 36 (73.5) | 0.033 |
| Married | 0 (0.0) | 4 (11.1) | 4 (8.2) | ||
| Separated or divorced | 0 (0.0) | 9 (25.0) | 9 (18.4) | ||
| Education | Junior high school and below | 1 (7.7) | 8 (22.2) | 9 (18.4) | 0.300 |
| High school | 2 (15.4) | 10 (27.8) | 12 (24.5) | ||
| College degree or above | 10 (76.9) | 18 (50.0) | 28 (57.1) | ||
| Occupation | Office worker | 9 (69.2) | 14 (38.9) | 23 (46.9) | 0.246 |
| Housework or unemployment | 2 (15.4) | 7 (19.4) | 9 (18.4) | ||
| Ordinary worker or farmer | 0 (0.0) | 5 (13.9) | 5 (10.2) | ||
| Sexual worker | 0 (0.0) | 6 (16.7) | 6 (12.2) | ||
| Student | 2 (15.4) | 4 (11.1) | 6 (12.2) | ||
| Subtype | CRF07_BC | 7 (53.8) | 20 (55.6) | 27 (55.1) | 0.031 |
| CRF55_01B | 0 (0.0) | 10 (27.8) | 10 (20.4) | ||
| CRF79_0107 | 6 (46.2) | 6 (16.7) | 12 (24.5) | ||
| Recency | Long-term infection | 10 (76.9) | 20 (55.6) | 30 (61.2) | 0.205 |
| Recent infection | 3 (23.1) | 16 (44.4) | 19 (38.8) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Cui, M.; Hong, Z.; Huang, S.; Zhou, S.; Lyu, H.; Li, J.; Lin, Y.; Huang, H.; Tang, W.; et al. High Genetic Diversity of HIV-1 and Active Transmission Clusters among Male-to-Male Sexual Contacts (MMSCs) in Zhuhai, China. Viruses 2023, 15, 1947. https://doi.org/10.3390/v15091947
Zhou Y, Cui M, Hong Z, Huang S, Zhou S, Lyu H, Li J, Lin Y, Huang H, Tang W, et al. High Genetic Diversity of HIV-1 and Active Transmission Clusters among Male-to-Male Sexual Contacts (MMSCs) in Zhuhai, China. Viruses. 2023; 15(9):1947. https://doi.org/10.3390/v15091947
Chicago/Turabian StyleZhou, Yi, Mingting Cui, Zhongsi Hong, Shaoli Huang, Shuntai Zhou, Hang Lyu, Jiarun Li, Yixiong Lin, Huitao Huang, Weiming Tang, and et al. 2023. "High Genetic Diversity of HIV-1 and Active Transmission Clusters among Male-to-Male Sexual Contacts (MMSCs) in Zhuhai, China" Viruses 15, no. 9: 1947. https://doi.org/10.3390/v15091947