

Xanthomonas Phage PBR31: Classifying the Unclassifiable

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
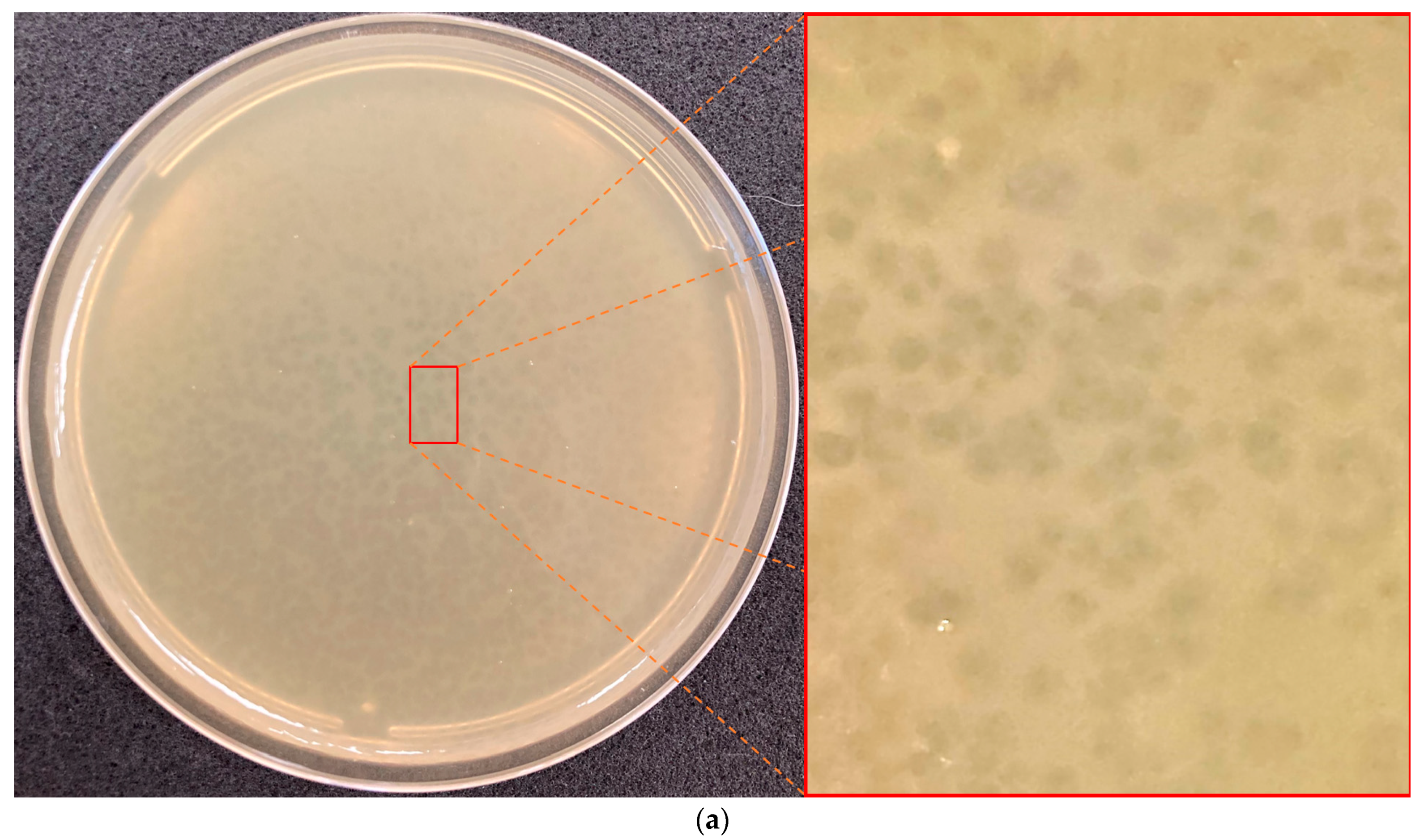
2.1. Bacteriophage Isolation and Purification
2.2. Determination of Phage Host Range
2.3. Phage Adsorption and One-Step Growth Experiments
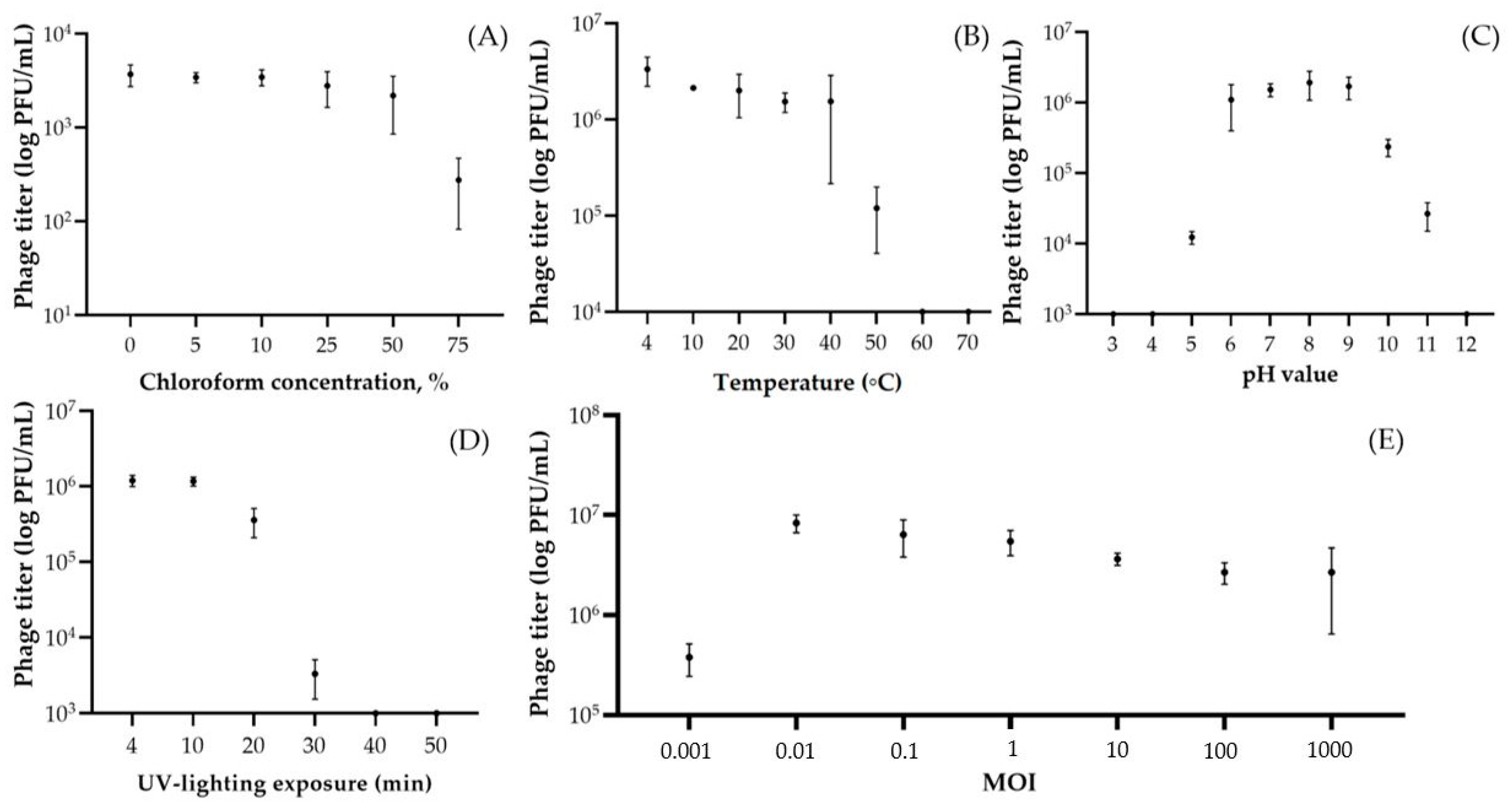
2.4. Phage Stability under Different Conditions
2.5. Calculation of MOI
2.6. Electron Microscopy
2.7. Phage Genome Sequencing and Annotation
2.8. Genome and Proteome Analysis
3. Results
3.1. Bacteriophage Isolation and Host Range Determination
3.2. Resistance to Stress Factors and Kinetic Features
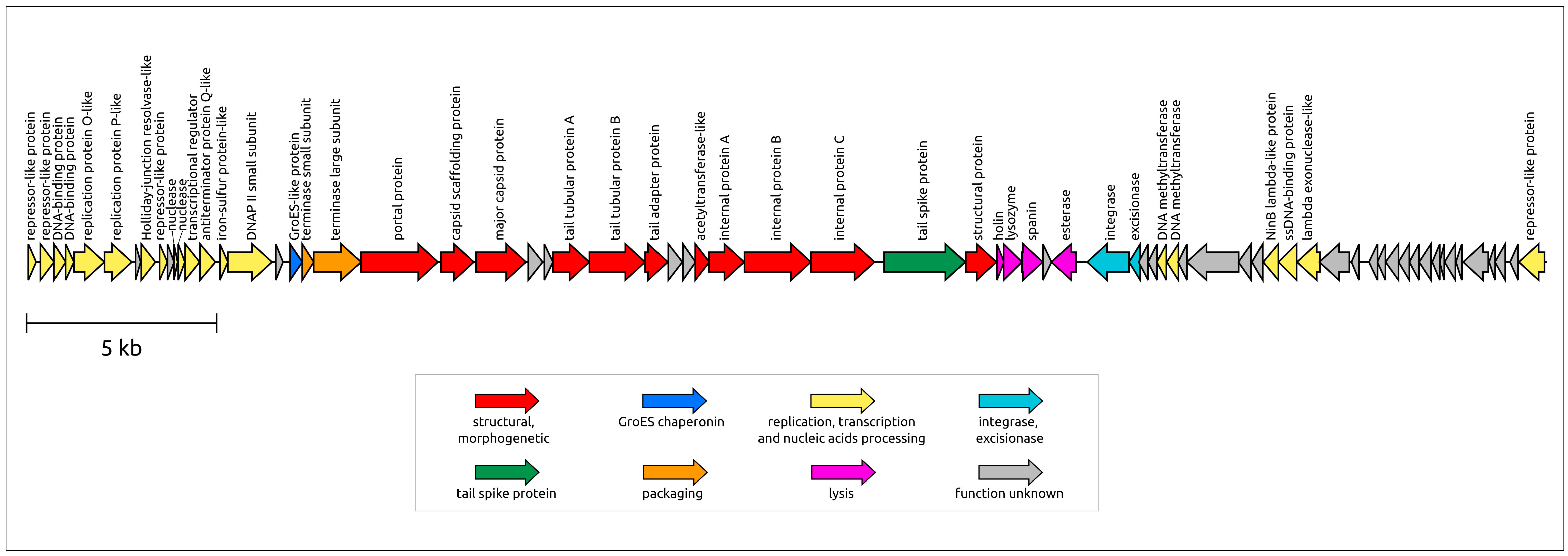
3.3. General Characterisation of the Genome
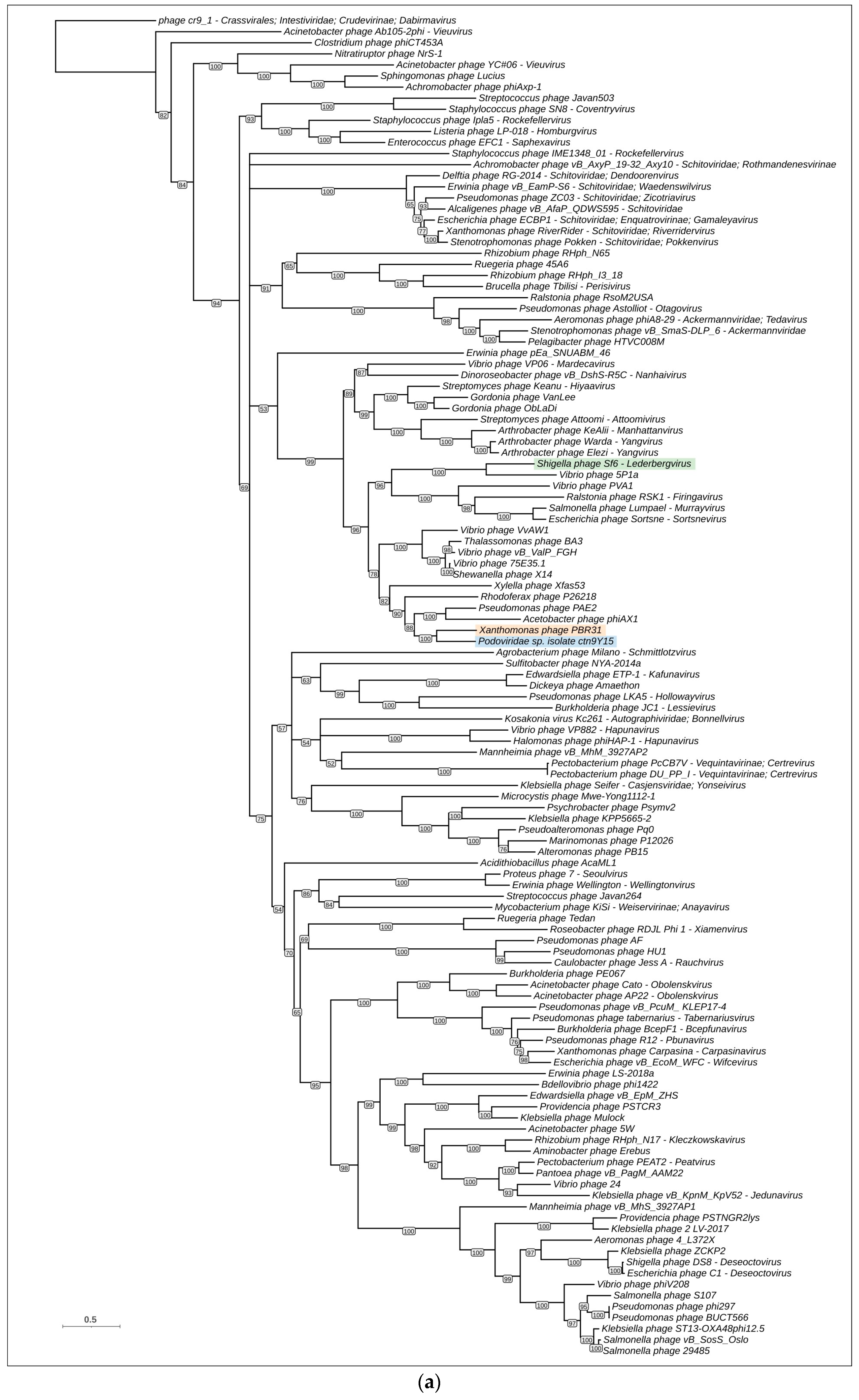
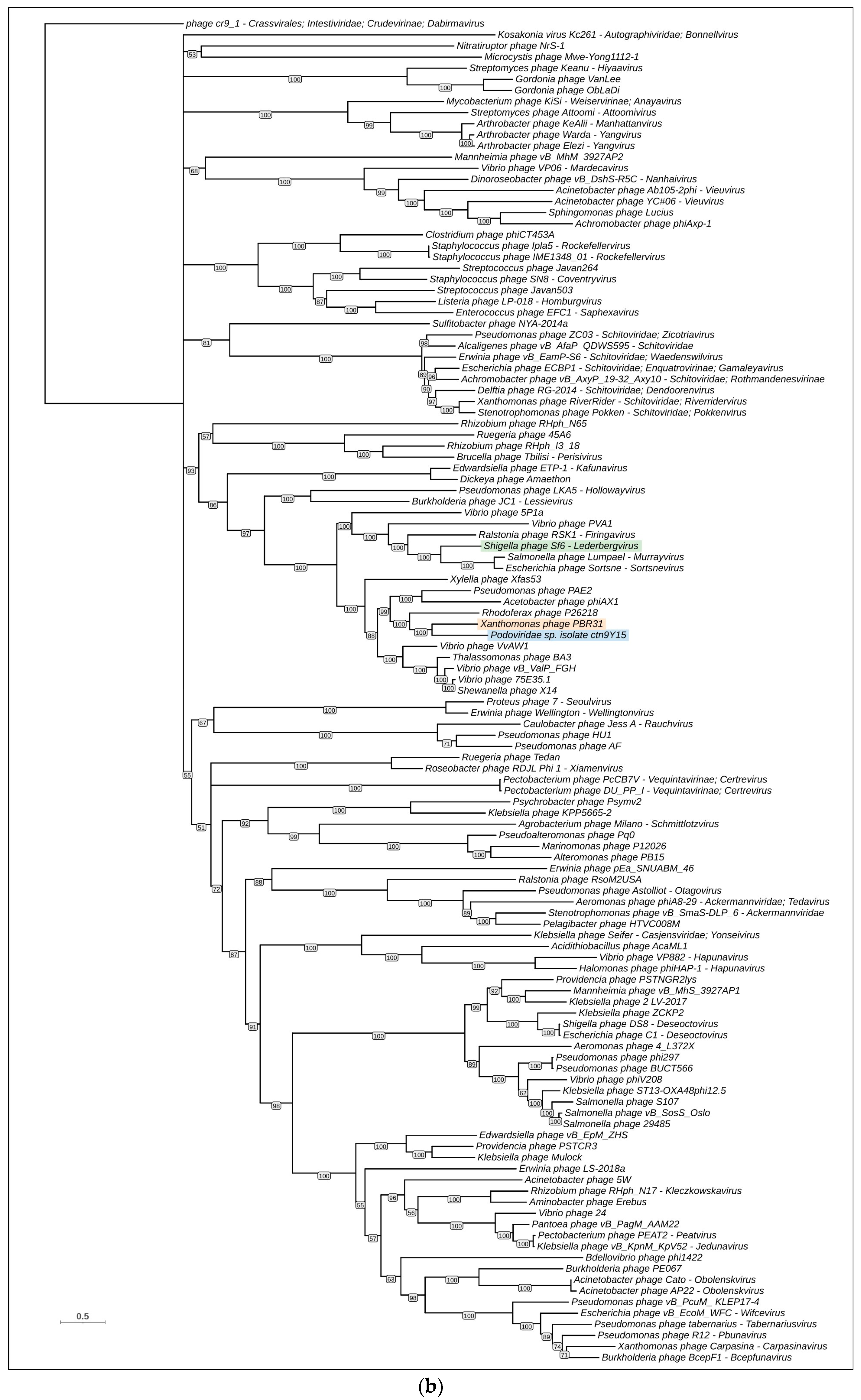
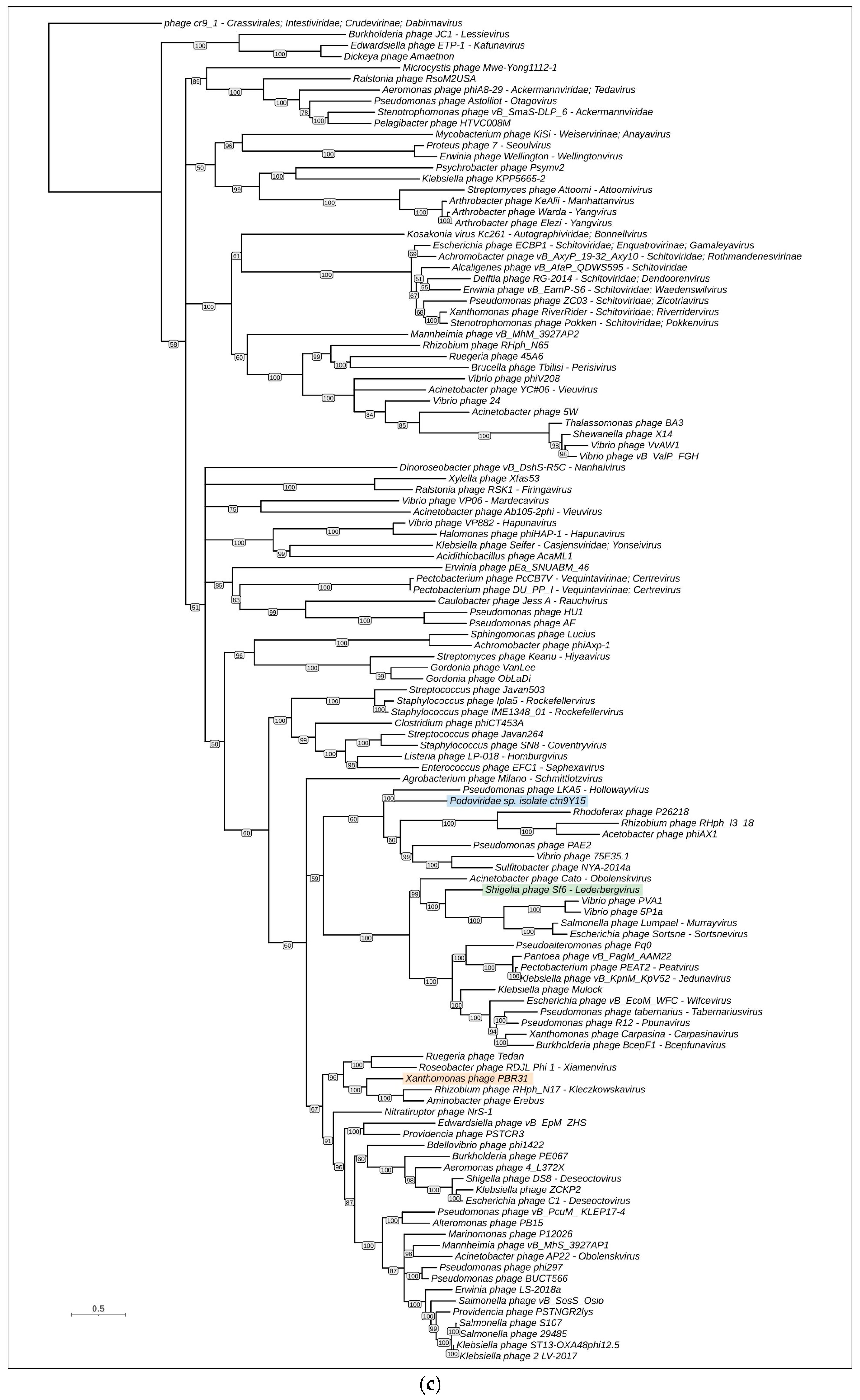
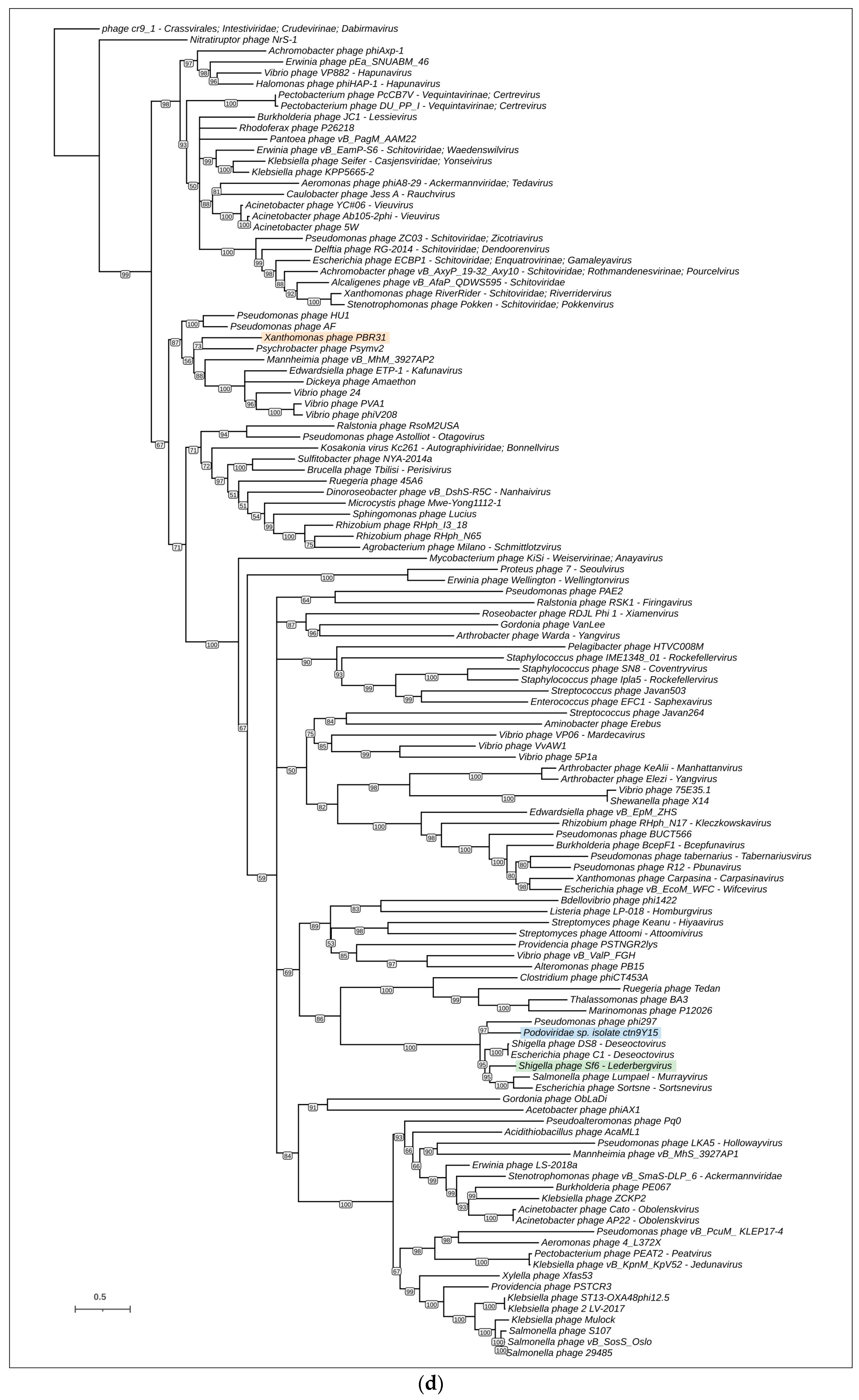
3.4. Phylogenetic and Phylogenomic Analyses
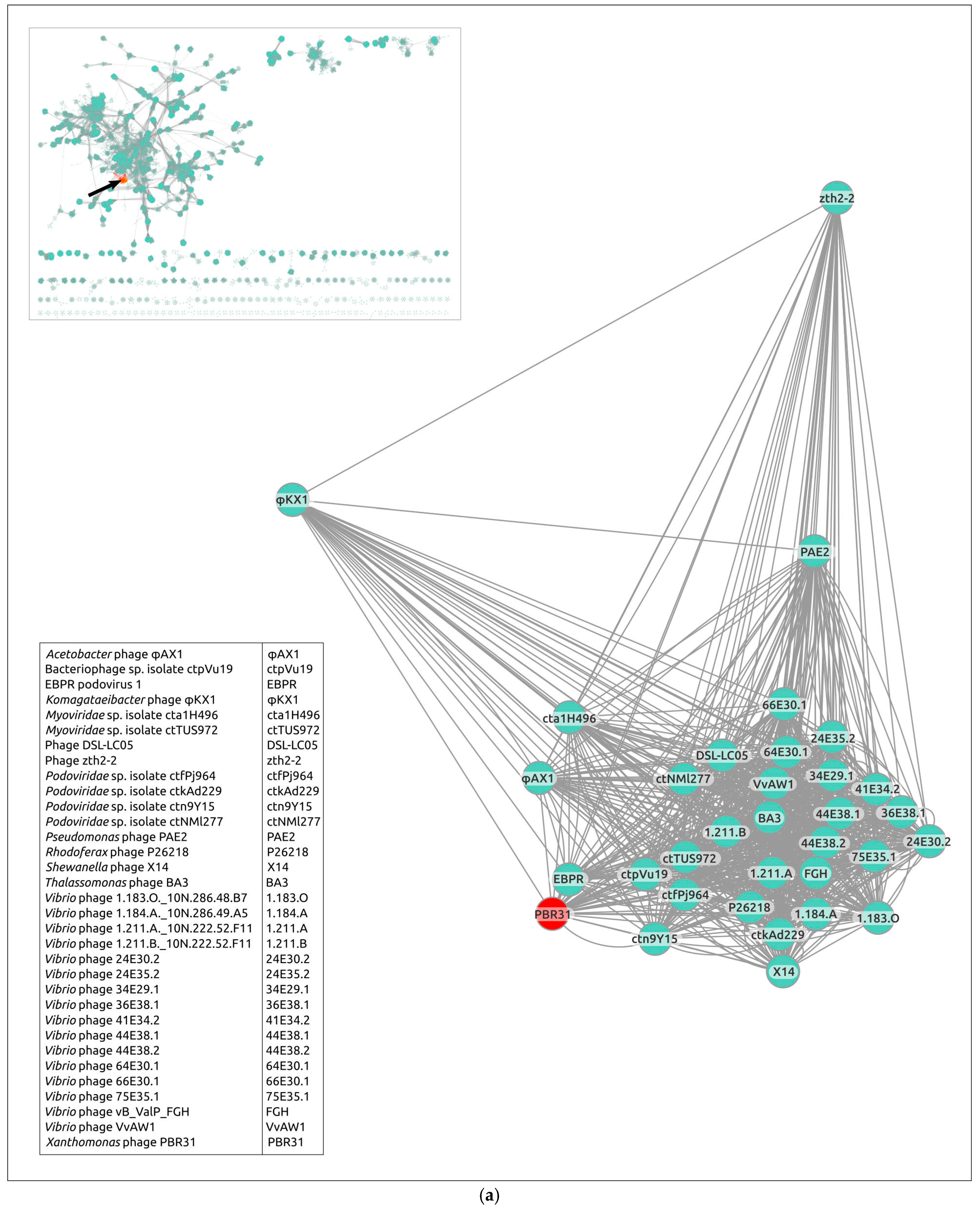
3.5. Gene Network Analysis and Intergenomic Comparisons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in Aquatic Ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef]
- Rohwer, F.; Youle, M.; Maughan, H.; Hisakawa, N. Life in Our Phage World: A Centennial Field Guide to the Earth’s Most Diverse Inhabitants; Wholon: San Diego, CA, USA, 2014; ISBN 978-0-9904943-0-0. [Google Scholar]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus Statement: Virus Taxonomy in the Age of Metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary Relationships among Diverse Bacteriophages and Prophages: All the World’s a Phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Loc-Carrillo, C.; Abedon, S.T. Pros and Cons of Phage Therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Bacteriophages. Virologie 2004, 8, 409–412. [Google Scholar]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to Virus Taxonomy and to the International Code of Virus Classification and Nomenclature Ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Curtis Hendrickson, R.; et al. Recent Changes to Virus Taxonomy Ratified by the International Committee on Taxonomy of Viruses (2022). Arch. Virol. 2022, 167, 2429–2440. [Google Scholar] [CrossRef]
- Zerbini, F.M.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Hendrickson, R.C.; et al. Changes to Virus Taxonomy and the ICTV Statutes Ratified by the International Committee on Taxonomy of Viruses (2023). Arch. Virol. 2023, 168, 175. [Google Scholar] [CrossRef]
- Low, S.J.; Džunková, M.; Chaumeil, P.-A.; Parks, D.H.; Hugenholtz, P. Evaluation of a Concatenated Protein Phylogeny for Classification of Tailed Double-Stranded DNA Viruses Belonging to the Order Caudovirales. Nat. Microbiol. 2019, 4, 1306–1315. [Google Scholar] [CrossRef]
- Yutin, N.; Wolf, Y.I.; Koonin, E.V. Origin of Giant Viruses from Smaller DNA Viruses Not from a Fourth Domain of Cellular Life. Virology 2014, 466, 38–52. [Google Scholar] [CrossRef]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origin of Viruses: Primordial Replicators Recruiting Capsids from Hosts. Nat. Rev. Microbiol. 2019, 17, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A. Comparative Molecular Biology of Lambdoid Phages. Annu. Rev. Microbiol. 1994, 48, 193–222. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Lukianova, A.; Sykilinda, N.; Gorshkova, A.; Bondar, A.; Shneider, M.; Kabilov, M.; Drucker, V.; Miroshnikov, K. Pseudomonas Phage MD8: Genetic Mosaicism and Challenges of Taxonomic Classification of Lambdoid Bacteriophages. Int. J. Mol. Sci. 2021, 22, 10350. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, D.; Fortuna, K.J.; Moons, L.; Broeckaert, N.; Bäcker, L.E.; Venneman, S.; Rombouts, S.; Lippens, L.; Baeyen, S.; Pollet, S.; et al. The Potential of Bacteriophages to Control Xanthomonas campestris pv. campestris at Different Stages of Disease Development. Microb. Biotechnol. 2022, 15, 1762–1782. [Google Scholar] [CrossRef]
- Williams, P.H. Black Rot: A Continuing Threat to World Crucifers. Plant Dis. 1980, 64, 736–742. [Google Scholar] [CrossRef]
- Monteiro, R.; Pires, D.P.; Costa, A.R.; Azeredo, J. Phage Therapy: Going Temperate? Trends Microbiol. 2019, 27, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A. Using Phage Lytic Enzymes to Control Pathogenic Bacteria. BMC Oral. Health 2006, 6 (Suppl. S1), S16. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.; Kenna, D.T.; Vandamme, P.; Ure, R.; Govan, J.R.W. Lysogeny and Bacteriophage Host Range within the Burkholderia cepacia Complex. J. Med. Microbiol. 2003, 52, 483–490. [Google Scholar] [CrossRef]
- Sambrook, J. Molecular Cloning: A Laboratory Manual, 2nd ed.; Sambrook, J., Fritsch, E.F., Maniatis, T., Eds.; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1989; ISBN 978-0-87969-309-1. [Google Scholar]
- Jäckel, C.; Hammerl, J.A.; Reetz, J.; Kropinski, A.M.; Hertwig, S. Campylobacter Group II Phage CP21 Is the Prototype of a New Subgroup Revealing a Distinct Modular Genome Organization and Host Specificity. BMC Genom. 2015, 16, 629. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, J.; Wang, Y.; Zhou, F.; Li, Q.; Teng, T. Phenotypic Characterization and Comparative Genomic Analyses of Mycobacteriophage WIVsmall as A New Member Assigned to F1 Subcluster. Curr. Issues Mol. Biol. 2023, 45, 6432–6448. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959. [Google Scholar]
- Wdowiak, M.; Paczesny, J.; Raza, S. Enhancing the Stability of Bacteriophages Using Physical, Chemical, and Nano-Based Approaches: A Review. Pharmaceutics 2022, 14, 1936. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Gao, Y.; Xue, Y.; Liu, Y.; Zeng, X.; Cheng, Y.; Ma, J.; Wang, H.; Sun, J.; Wang, Z.; et al. Bacteriophage Cocktails Protect Dairy Cows Against Mastitis Caused By Drug Resistant Escherichia coli Infection. Front. Cell Infect. Microbiol. 2021, 11, 690377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ding, Y.; Li, W.; Zhu, W.; Wang, J.; Wang, X. Application of a Novel Lytic Podoviridae Phage Pu20 for Biological Control of Drug-Resistant Salmonella in Liquid Eggs. Pathogens 2021, 10, 34. [Google Scholar] [CrossRef]
- Bouras, G.; Nepal, R.; Houtak, G.; Psaltis, A.J.; Wormald, P.-J.; Vreugde, S. Pharokka: A Fast Scalable Bacteriophage Annotation Tool. Bioinformatics 2023, 39, btac776. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a Program to Detect tRNA Genes and tmRNA Genes in Nucleotide Sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap.Js: Automatic Generation of Gene Cluster Comparison Figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The Viral Proteomic Tree Server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Dali Server: Structural Unification of Protein Families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.-Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus Tool to Classify Double-Stranded DNA Viruses That Infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D.J.; Hobman, J.; Jones, M.A.; Millard, A. INfrastructure for a PHAge REference Database: Identification of Large-Scale Biases in the Current Collection of Cultured Phage Genomes. Phage 2021, 2, 214–223. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kemp, P.; Garcia, L.R.; Molineux, I.J. Changes in Bacteriophage T7 Virion Structure at the Initiation of Infection. Virology 2005, 340, 307–317. [Google Scholar] [CrossRef]
- ASM Division M: Bacteriophage P22. Available online: https://web.archive.org/web/20101226170811/http://www.asm.org/division/m/fax/P22Fax.html (accessed on 21 February 2024).
- Zylicz, M.; Liberek, K.; Wawrzynow, A.; Georgopoulos, C. Formation of the Preprimosome Protects λ O from RNA Transcription-Dependent Proteolysis by ClpP/ClpX. Proc. Natl. Acad. Sci. USA 1998, 95, 15259–15263. [Google Scholar] [CrossRef] [PubMed]
- Morcinek-Orłowska, J.; Zdrojewska, K.; Węgrzyn, A. Bacteriophage-Encoded DNA Polymerases—Beyond the Traditional View of Polymerase Activities. Int. J. Mol. Sci. 2022, 23, 635. [Google Scholar] [CrossRef]
- Ravin, V.; Ravin, N.; Casjens, S.; Ford, M.E.; Hatfull, G.F.; Hendrix, R.W. Genomic Sequence and Analysis of the Atypical Temperate Bacteriophage N15. J. Mol. Biol. 2000, 299, 53–73. [Google Scholar] [CrossRef]
- van der Wilk, F.; Dullemans, A.M.; Verbeek, M.; van den Heuvel, J.F. Isolation and Characterization of APSE-1, a Bacteriophage Infecting the Secondary Endosymbiont of Acyrthosiphon pisum. Virology 1999, 262, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Mayanagi, K.; Oki, K.; Miyazaki, N.; Ishino, S.; Yamagami, T.; Morikawa, K.; Iwasaki, K.; Kohda, D.; Shirai, T.; Ishino, Y. Two Conformations of DNA Polymerase D-PCNA-DNA, an Archaeal Replisome Complex, Revealed by Cryo-Electron Microscopy. BMC Biol. 2020, 18, 152. [Google Scholar] [CrossRef] [PubMed]
- Honarmand Ebrahimi, K.; Ciofi-Baffoni, S.; Hagedoorn, P.-L.; Nicolet, Y.; Le Brun, N.E.; Hagen, W.R.; Armstrong, F.A. Iron–Sulfur Clusters as Inhibitors and Catalysts of Viral Replication. Nat. Chem. 2022, 14, 253–266. [Google Scholar] [CrossRef]
- Cann, I.K.; Ishino, Y. Archaeal DNA Replication: Identifying the Pieces to Solve a Puzzle. Genetics 1999, 152, 1249–1267. [Google Scholar] [CrossRef]
- Hilbert, B.J.; Hayes, J.A.; Stone, N.P.; Xu, R.-G.; Kelch, B.A. The Large Terminase DNA Packaging Motor Grips DNA with Its ATPase Domain for Cleavage by the Flexible Nuclease Domain. Nucleic Acids Res. 2017, 45, 3591–3605. [Google Scholar] [CrossRef]
- Gaïa, M.; Meng, L.; Pelletier, E.; Forterre, P.; Vanni, C.; Fernandez-Guerra, A.; Jaillon, O.; Wincker, P.; Ogata, H.; Krupovic, M.; et al. Mirusviruses Link Herpesviruses to Giant Viruses. Nature 2023, 616, 783–789. [Google Scholar] [CrossRef]
- Steinbacher, S.; Miller, S.; Baxa, U.; Budisa, N.; Weintraub, A.; Seckler, R.; Huber, R. Phage P22 Tailspike Protein: Crystal Structure of the Head-Binding Domain at 2.3 Å, Fully Refined Structure of the Endorhamnosidase at 1.56 Å Resolution, and the Molecular Basis of O-Antigen Recognition and Cleavage. J. Mol. Biol. 1997, 267, 865–880. [Google Scholar] [CrossRef]
- Liang, L.; Zhao, H.; An, B.; Tang, L. High-Resolution Structure of Podovirus Tail Adaptor Suggests Repositioning of an Octad Motif That Mediates the Sequential Tail Assembly. Proc. Natl. Acad. Sci. USA 2018, 115, 313–318. [Google Scholar] [CrossRef]
- Gage, M.J.; Robinson, A.S. C-Terminal Hydrophobic Interactions Play a Critical Role in Oligomeric Assembly of the P22 Tailspike Trimer. Protein Sci. 2003, 12, 2732–2747. [Google Scholar] [CrossRef]
- Duda, R.L.; Oh, B.; Hendrix, R.W. Functional Domains of the HK97 Capsid Maturation Protease and the Mechanisms of Protein Encapsidation. J. Mol. Biol. 2013, 425, 2765–2781. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Phage Lysis: Three Steps, Three Choices, One Outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Kongari, R.; Rajaure, M.; Cahill, J.; Rasche, E.; Mijalis, E.; Berry, J.; Young, R. Phage Spanins: Diversity, Topological Dynamics and Gene Convergence. BMC Bioinform. 2018, 19, 326. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.B.; Ray, K.; Thomas, J.A.; Black, L.W. The C-Terminal Domain of the Bacteriophage T4 Terminase Docks on the Prohead Portal Clip Region during DNA Packaging. Virology 2013, 446, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, B.; Monroe, L.; Li, K.; Yan, R.; Wright, E.; Walter, T.; Kihara, D.; Weintraub, S.T.; Thomas, J.A.; Serwer, P.; et al. Phage G Structure at 6.1 Å Resolution, Condensed DNA, and Host Identity Revision to a Lysinibacillus. J. Mol. Biol. 2020, 432, 4139–4153. [Google Scholar] [CrossRef]
- Lokareddy, R.K.; Hou, C.-F.D.; Doll, S.G.; Li, F.; Gillilan, R.E.; Forti, F.; Horner, D.S.; Briani, F.; Cingolani, G. Terminase Subunits from the Pseudomonas-Phage E217. J. Mol. Biol. 2022, 434, 167799. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D. A Theory of Modular Evolution for Bacteriophages. Ann. N. Y. Acad. Sci. 1980, 354, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Omata, K.; Hibi, N.; Nakano, S.; Komoto, S.; Sato, K.; Nunokawa, K.; Amano, S.; Ueda, K.; Takano, H. Distribution and Genome Structures of Temperate Phages in Acetic Acid Bacteria. Sci. Rep. 2021, 11, 21567. [Google Scholar] [CrossRef]
- Qian, Z.; Hui, P.; Han, L.; Yang, L.-Z.; Zhang, B.-S.; Jie, Z.; Guo, W.-L.; Nan, W.; Jiang, S.-J.; Xie, Z.-J. Serotypes and Virulence Genes of Pseudomonas aeruginosa Isolated from Mink and Its Pathogenicity in Mink. Microb. Pathog. 2020, 139, 103904. [Google Scholar] [CrossRef]
- Moon, K.; Kang, I.; Kim, S.; Cho, J.-C.; Kim, S.-J. Complete Genome Sequence of Bacteriophage P26218 Infecting Rhodoferax sp. Strain IMCC26218. Stand. Genom. Sci. 2015, 10, 111. [Google Scholar] [CrossRef]
- Summer, E.J.; Enderle, C.J.; Ahern, S.J.; Gill, J.J.; Torres, C.P.; Appel, D.N.; Black, M.C.; Young, R.; Gonzalez, C.F. Genomic and Biological Analysis of Phage Xfas53 and Related Prophages of Xylella fastidiosa. J. Bacteriol. 2010, 192, 179–190. [Google Scholar] [CrossRef]
- Nigro, O.D.; Culley, A.I.; Steward, G.F. Complete Genome Sequence of Bacteriophage VvAW1, Which Infects Vibrio vulnificus. Stand. Genom. Sci. 2012, 6, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Dover, J.A.; Parent, K.N.; Doore, S.M. Host Range Expansion of Shigella Phage Sf6 Evolves through Point Mutations in the Tailspike. J. Virol. 2022, 96, e00929-22. [Google Scholar] [CrossRef]
- Trotereau, A.; Boyer, C.; Bornard, I.; Pécheur, M.J.B.; Schouler, C.; Torres-Barceló, C. High Genomic Diversity of Novel Phages Infecting the Plant Pathogen Ralstonia solanacearum, Isolated in Mauritius and Reunion Islands. Sci. Rep. 2021, 11, 5382. [Google Scholar] [CrossRef]
- Meiring, T.L.; Tuffin, I.M.; Cary, C.; Cowan, D.A. Genome Sequence of Temperate Bacteriophage Psymv2 from Antarctic Dry Valley Soil Isolate Psychrobacter sp. MV2. Extremophiles 2012, 16, 715–726. [Google Scholar] [CrossRef]
- Evseev, P.; Gutnik, D.; Shneider, M.; Miroshnikov, K. Use of an Integrated Approach Involving AlphaFold Predictions for the Evolutionary Taxonomy of Duplodnaviria Viruses. Biomolecules 2023, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Nakayinga, R.; Makumi, A.; Tumuhaise, V.; Tinzaara, W. Xanthomonas Bacteriophages: A Review of Their Biology and Biocontrol Applications in Agriculture. BMC Microbiol. 2021, 21, 291. [Google Scholar] [CrossRef] [PubMed]
- Penadés, J.R.; Chen, J.; Quiles-Puchalt, N.; Carpena, N.; Novick, R.P. Bacteriophage-Mediated Spread of Bacterial Virulence Genes. Curr. Opin. Microbiol. 2015, 23, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.R.; Allison, H.; James, C.E.; McCarthy, A.J.; Sharp, R. Phage-Mediated Transfer of Virulence Genes. J. Chem. Technol. Biotechnol. 2001, 76, 662–666. [Google Scholar] [CrossRef]
- Allison, H.E. Stx-Phages: Drivers and Mediators of the Evolution of STEC and STEC-like Pathogens. Future Microbiol. 2007, 2, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Portrait of a Viral Infection: The Infection Cycle of Vibrio vulnificus Phage VvAW1 Visualized through Plaque Assay, Electron Microscopy, and Proteomics—NASA/ADS. Available online: https://ui.adsabs.harvard.edu/abs/2016AGUOSMM44D0522C/abstract (accessed on 22 October 2023).
- Owen, S.V.; Wenner, N.; Canals, R.; Makumi, A.; Hammarlöf, D.L.; Gordon, M.A.; Aertsen, A.; Feasey, N.A.; Hinton, J.C.D. Characterization of the Prophage Repertoire of African Salmonella typhimurium ST313 Reveals High Levels of Spontaneous Induction of Novel Phage BTP1. Front. Microbiol. 2017, 8, 235. [Google Scholar] [CrossRef]
- Pas, C.; Latka, A.; Fieseler, L.; Briers, Y. Phage Tailspike Modularity and Horizontal Gene Transfer Reveals Specificity towards E. coli O-Antigen Serogroups. Virol. J. 2023, 20, 174. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Multiple Origins of Viral Capsid Proteins from Cellular Ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Strain | Date of Isolation | Place of Origin, Plant | Genbank 16S № | Lysis Zone Caused by Phage R3-1 |
|---|---|---|---|---|
| BK-55 | 10.2017 | Krasnodar region, Russia, white cabbage | OR626094 | + |
| CK-71 | 10.2017 | Krasnodar region, Russia, cauliflower | OR626097 | + |
| Xcc 1/1 | 09.2017 | Moscow region, Dmitrov, Russia, white cabbage | OR626648 | + |
| Bes-1 | 09.2016 | Moscow region, Dmitrov, Russia, white cabbage | OR626092 | - |
| Cas | 09.2016 | Moscow region, Dmitrov, Russia, cauliflower | OR626095 | + |
| Tr1 | 11.2012 | Tiraspol, Transnistria, Moldova, cabbage | OR626099 | + |
| DK-1 | 10.2012 | Moscow region, Serpukhov, Russia, white cabbage | OR626096 | + |
| Ram 3-1 | 10.2012 | Moscow region, Ramensky, Russia, cabbage | OR625211 | + |
| XУ 1-2 | 10.2012 | Ukraine, white cabbage | OR644606 | - |
| Bel-2 | 10.2006 | Belarus, white cabbage | OR626091 | - |
| Bun-1 | 09.2006 | Moscow region, Dmitrov, Russia, white cabbage | OR626093 | + |
| Xn-13 | 1997 | Japan, Mie-ken, Capsélla búrsa-pastóris (shepherd’s purse) | OR626098 | + |
| 306NZ | - | The Netherlands | OR626090 | + |
| NCPPB 528T | 1957 | UK, cabbage | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarakanov, R.I.; Evseev, P.V.; Vo, H.T.N.; Troshin, K.S.; Gutnik, D.I.; Ignatov, A.N.; Toshchakov, S.V.; Miroshnikov, K.A.; Jafarov, I.H.; Dzhalilov, F.S.-U. Xanthomonas Phage PBR31: Classifying the Unclassifiable. Viruses 2024, 16, 406. https://doi.org/10.3390/v16030406
Tarakanov RI, Evseev PV, Vo HTN, Troshin KS, Gutnik DI, Ignatov AN, Toshchakov SV, Miroshnikov KA, Jafarov IH, Dzhalilov FS-U. Xanthomonas Phage PBR31: Classifying the Unclassifiable. Viruses. 2024; 16(3):406. https://doi.org/10.3390/v16030406
Chicago/Turabian StyleTarakanov, Rashit I., Peter V. Evseev, Ha T. N. Vo, Konstantin S. Troshin, Daria I. Gutnik, Aleksandr N. Ignatov, Stepan V. Toshchakov, Konstantin A. Miroshnikov, Ibrahim H. Jafarov, and Fevzi S.-U. Dzhalilov. 2024. "Xanthomonas Phage PBR31: Classifying the Unclassifiable" Viruses 16, no. 3: 406. https://doi.org/10.3390/v16030406