
Pathogenic and Apathogenic Strains of Lymphocytic Choriomeningitis Virus Have Distinct Entry and Innate Immune Activation Pathways

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Treatment with Chemical Inhibitors
2.3. IRF and NF-κB Activation in Dual-Reporter Cell Lines
2.4. TLR-2 Silencing in LCMV-Infected Cells
2.5. Immunofluorescence Co-Staining, Confocal Microscopy
2.6. Western Blot Analysis of IRAK-1 Expression
2.7. Statistical Analyses
3. Results
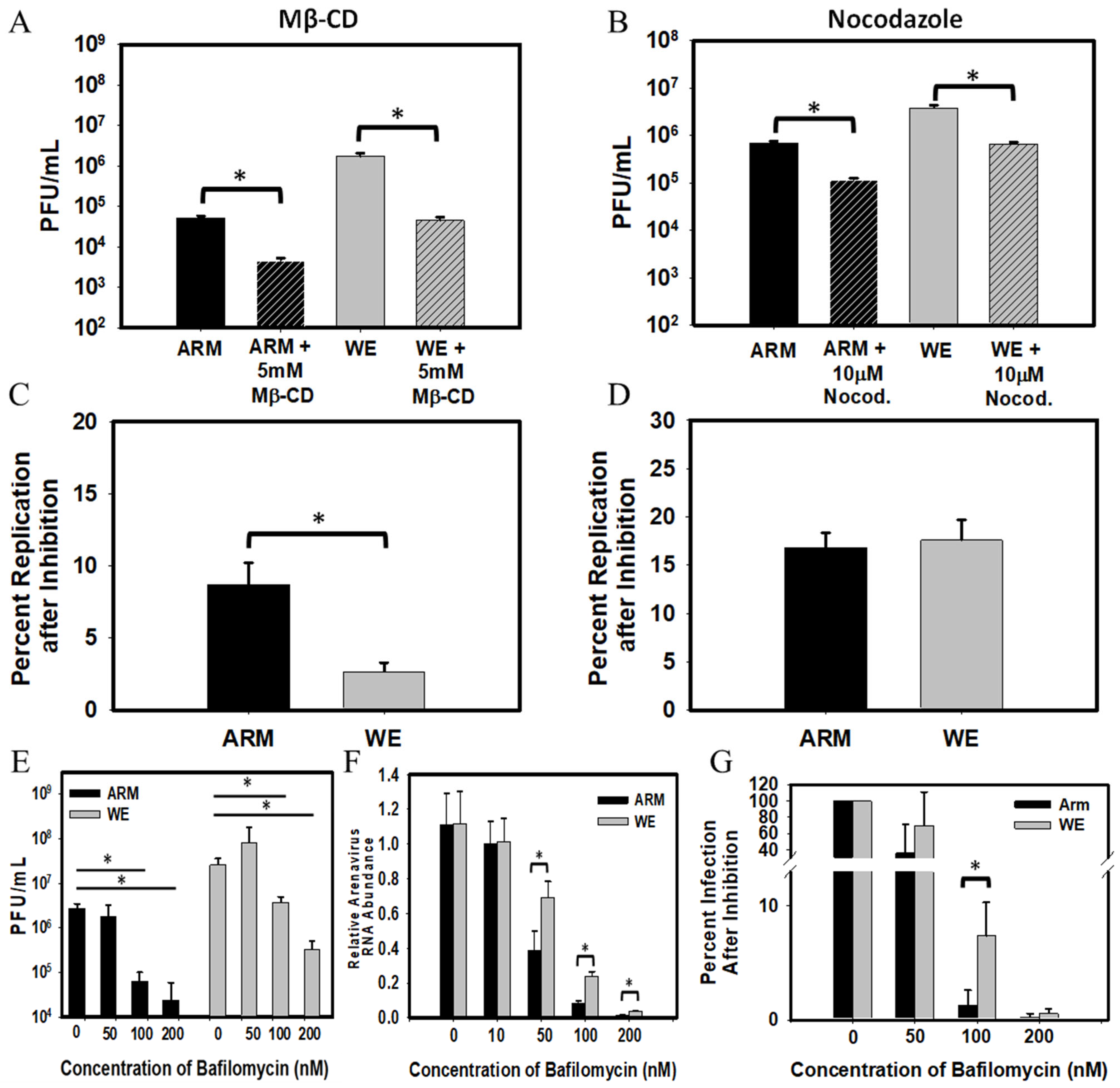
3.1. Probing LCMV-ARM versus LCMV-WE Entry with Chemical Inhibitors
3.2. Innate Immune Activation in Cells Infected with LCMV-ARM versus LCMV-WE

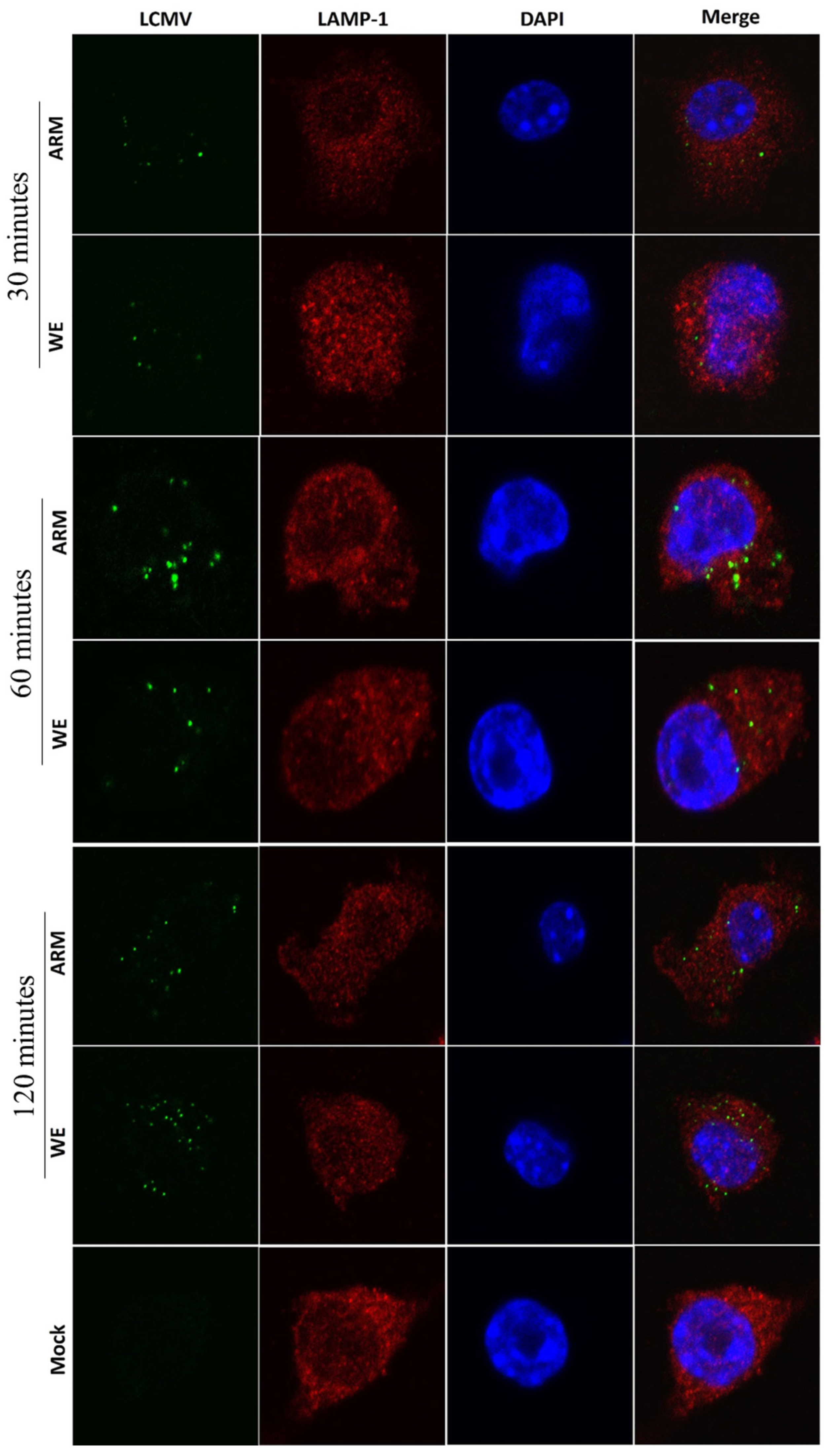
3.3. Co-Staining Experiments with Markers of LCMV Infection, TLR-2, and Early Endosomes
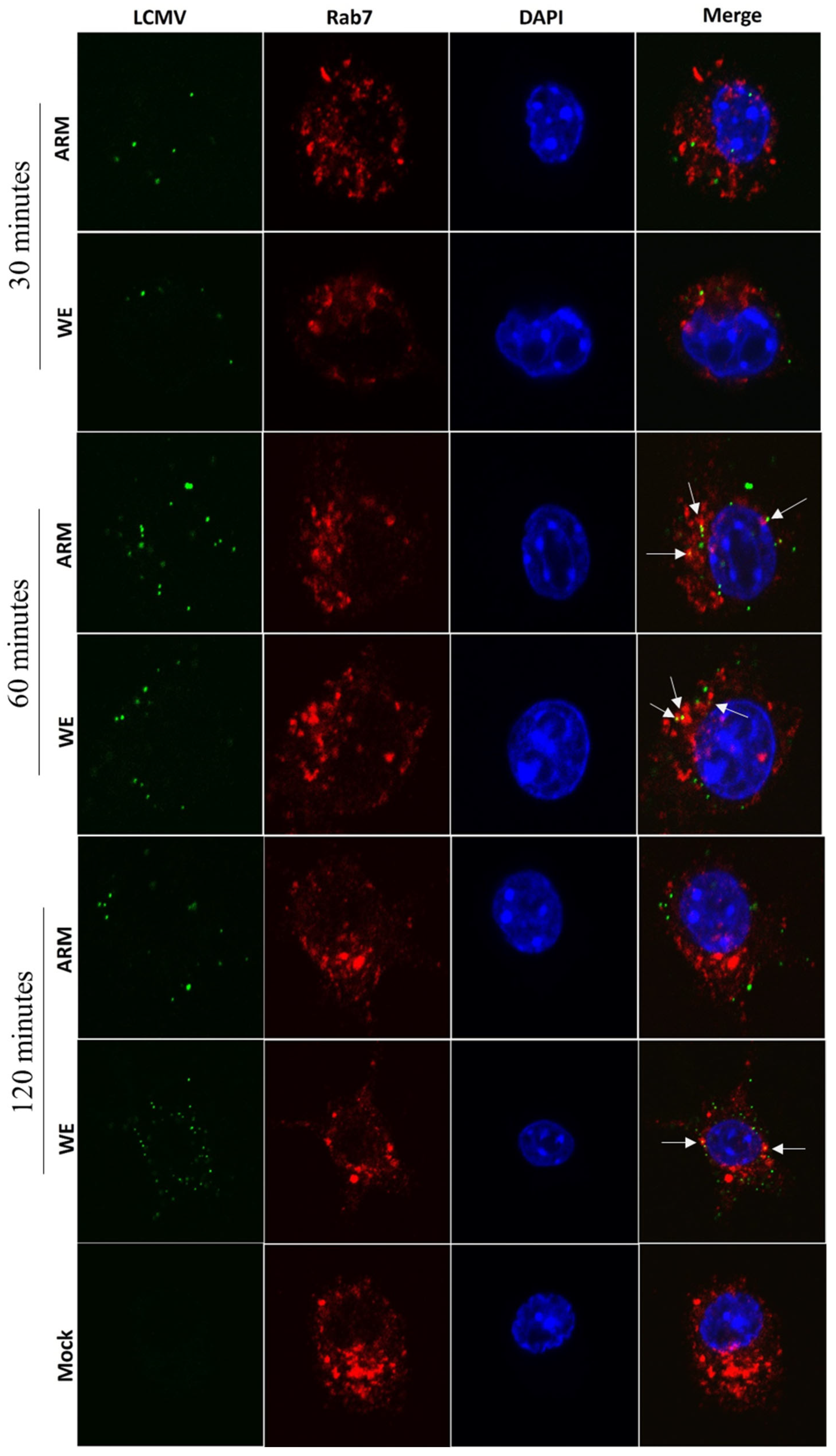
3.4. LCMV Interaction with Markers of Late Endosomes/Exosomes

3.5. Interaction with IRAK-1, Mediator of TLR-Induced Signaling, in LCMV-Infected Cells

4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonthius, D.J. Lymphocytic Choriomeningitis Virus: An Underrecognized Cause of Neurologic Disease in the Fetus, Child, and Adult. In Seminars in Pediatric Neurology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 19, pp. 89–95. [Google Scholar]
- Lapošová, K.; Pastoreková, S.; Tomášková, J. Lymphocytic Choriomeningitis Virus: Invisible but Not Innocent. Acta Virol. 2013, 57, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Radoshitzky, S.R.; Buchmeier, M.J.; Charrel, R.N.; Gonzalez, J.-P.J.; Günther, S.; Hepojoki, J.; Kuhn, J.H.; Lukashevich, I.S.; Romanowski, V.; Salvato, M.S. ICTV Virus Taxonomy Profile: Arenaviridae 2023. J. Gen. Virol. 2023, 104, 001891. [Google Scholar] [CrossRef]
- Price, M.E.; Fisher-Hoch, S.P.; Craven, R.B.; McCormick, J.B. A Prospective Study of Maternal and Fetal Outcome in Acute Lassa Fever Infection during Pregnancy. Br. Med. J. 1988, 297, 584–587. [Google Scholar] [CrossRef]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B. Identification of α-Dystroglycan as a Receptor for Lymphocytic Choriomeningitis Virus and Lassa Fever Virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef]
- Peters, C.J.; Jahrling, P.B.; Liu, C.T.; Kenyon, R.H.; McKee, K.T.; Barrera Oro, J.G. Experimental Studies of Arenaviral Hemorrhagic Fevers. In Arenaviruses: Biology and Immunotherapy; Springer: Berlin/Heidelberg, Germany, 1987; pp. 5–68. ISBN 3-642-71728-4. [Google Scholar]
- Zapata, J.C.; Pauza, C.D.; Djavani, M.M.; Rodas, J.D.; Moshkoff, D.; Bryant, J.; Ateh, E.; Garcia, C.; Lukashevich, I.S.; Salvato, M.S. Lymphocytic Choriomeningitis Virus (LCMV) Infection of Macaques: A Model for Lassa Fever. Antivir. Res. 2011, 92, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Albariño, C.G.; Palacios, G.; Khristova, M.L.; Erickson, B.R.; Carroll, S.A.; Comer, J.A.; Hui, J.; Briese, T.; George, K.S.; Ksiazek, T.G. High Diversity and Ancient Common Ancestry of Lymphocytic Choriomeningitis Virus. Emerg. Infect. Dis. 2010, 16, 1093–1100. [Google Scholar] [CrossRef]
- Arruda, L.B.; Haider, N.; Olayemi, A.; Simons, D.; Ehichioya, D.; Yinka-Ogunleye, A.; Ansumana, R.; Thomason, M.J.; Asogun, D.; Ihekweazu, C. The Niche of One Health Approaches in Lassa Fever Surveillance and Control. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- Bowen, M.D.; Rollin, P.E.; Ksiazek, T.G.; Hustad, H.L.; Bausch, D.G.; Demby, A.H.; Bajani, M.D.; Peters, C.J.; Nichol, S.T. Genetic Diversity among Lassa Virus Strains. J. Virol. 2000, 74, 6992–7004. [Google Scholar] [CrossRef]
- Kouadio, L.; Nowak, K.; Akoua-Koffi, C.; Weiss, S.; Allali, B.K.; Witkowski, P.T.; Krüger, D.H.; Couacy-Hymann, E.; Calvignac-Spencer, S.; Leendertz, F.H. Lassa Virus in Multimammate Rats, Côte d’Ivoire, 2013. Emerg. Infect. Dis. 2015, 21, 1481–1483. [Google Scholar] [CrossRef]
- Manning, J.T.; Forrester, N.; Paessler, S. Lassa Virus Isolates from Mali and the Ivory Coast Represent an Emerging Fifth Lineage. Front. Microbiol. 2015, 6, 1037. [Google Scholar] [CrossRef]
- Safronetz, D.; Lopez, J.E.; Sogoba, N.; Traore, S.F.; Raffel, S.J.; Fischer, E.R.; Ebihara, H.; Branco, L.; Garry, R.F.; Schwan, T.G. Detection of Lassa Virus, Mali. Emerg. Infect. Dis. 2010, 16, 1123–1126. [Google Scholar] [CrossRef]
- Whitmer, S.L.; Strecker, T.; Cadar, D.; Dienes, H.-P.; Faber, K.; Patel, K.; Brown, S.M.; Davis, W.G.; Klena, J.D.; Rollin, P.E. New Lineage of Lassa Virus, Togo, 2016. Emerg. Infect. Dis. 2018, 24, 599–602. [Google Scholar] [CrossRef]
- McCormick, J.B.; Webb, P.A.; Krebs, J.W.; Johnson, K.M.; Smith, E.S. A Prospective Study of the Epidemiology and Ecology of Lassa Fever. J. Infect. Dis. 1987, 155, 437–444. [Google Scholar] [CrossRef]
- Garry, R.F. Lassa Fever—The Road Ahead. Nat. Rev. Microbiol. 2023, 21, 87–96. [Google Scholar] [CrossRef]
- Fischer, S.A.; Graham, M.B.; Kuehnert, M.J.; Kotton, C.N.; Srinivasan, A.; Marty, F.M.; Comer, J.A.; Guarner, J.; Paddock, C.D.; DeMeo, D.L. Transmission of Lymphocytic Choriomeningitis Virus by Organ Transplantation. N. Engl. J. Med. 2006, 354, 2235–2249. [Google Scholar] [CrossRef]
- MacNeil, A.; Ströher, U.; Farnon, E.; Campbell, S.; Cannon, D.; Paddock, C.D.; Drew, C.P.; Kuehnert, M.; Knust, B.; Gruenenfelder, R. Solid Organ Transplant–Associated Lymphocytic Choriomeningitis, United States, 2011. Emerg. Infect. Dis. 2012, 18, 1256–1262. [Google Scholar] [CrossRef]
- Zinkernagel, R.M.; Haenseler, E.; Leist, T.; Cerny, A.; Hengartner, H.; Althage, A. T Cell-Mediated Hepatitis in Mice Infected with Lymphocytic Choriomeningitis Virus. Liver Cell Destruction by H-2 Class I-Restricted Virus-Specific Cytotoxic T Cells as a Physiological Correlate of the 51Cr-Release Assay? J. Exp. Med. 1986, 164, 1075–1092. [Google Scholar] [CrossRef]
- Lang, P.A.; Recher, M.; Honke, N.; Scheu, S.; Borkens, S.; Gailus, N.; Krings, C.; Meryk, A.; Kulawik, A.; Cervantes-Barragan, L. Tissue Macrophages Suppress Viral Replication and Prevent Severe Immunopathology in an interferon-I-dependent Manner in Mice. Hepatology 2010, 52, 25–32. [Google Scholar] [CrossRef]
- Beier, J.I.; Jokinen, J.D.; Holz, G.E.; Whang, P.S.; Martin, A.M.; Warner, N.L.; Arteel, G.E.; Lukashevich, I.S. Novel Mechanism of Arenavirus-Induced Liver Pathology. PLoS ONE 2015, 10, e0122839. [Google Scholar] [CrossRef]
- Oldstone, M.B. Principles of Viral Pathogenesis. Cell 1996, 87, 799–801. [Google Scholar] [CrossRef]
- Matullo, C.M.; O’Regan, K.J.; Hensley, H.; Curtis, M.; Rall, G.F. Lymphocytic Choriomeningitis Virus-Induced Mortality in Mice Is Triggered by Edema and Brain Herniation. J. Virol. 2010, 84, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Ware, B.C.; Sullivan, B.M.; LaVergne, S.; Marro, B.S.; Egashira, T.; Campbell, K.P.; Elder, J.; Oldstone, M.B. A Unique Variant of Lymphocytic Choriomeningitis Virus That Induces Pheromone Binding Protein MUP: Critical Role for CTL. Proc. Natl. Acad. Sci. USA 2019, 116, 18001–18008. [Google Scholar] [CrossRef]
- Sevilla, N.; Domingo, E.; de la Torre, J.C. Contribution of LCMV towards Deciphering Biology of Quasispecies in Vivo. Arenaviruses II Mol. Pathog. Arenavirus Infect. 2002, 263, 197–220. [Google Scholar]
- Sevilla, N.; Kunz, S.; Holz, A.; Lewicki, H.; Homann, D.; Yamada, H.; Campbell, K.P.; de la Torre, J.C.; Oldstone, M.B. Immunosuppression and Resultant Viral Persistence by Specific Viral Targeting of Dendritic Cells. J. Exp. Med. 2000, 192, 1249–1260. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Tikhonov, I.; Rodas, J.D.; Zapata, J.C.; Yang, Y.; Djavani, M.; Salvato, M.S. Arenavirus-Mediated Liver Pathology: Acute Lymphocytic Choriomeningitis Virus Infection of Rhesus Macaques Is Characterized by High-Level Interleukin-6 Expression and Hepatocyte Proliferation. J. Virol. 2003, 77, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S.; Rodas, J.D.; Tikhonov, I.I.; Zapata, J.C.; Yang, Y.; Djavani, M.; Salvato, M.S. LCMV-Mediated Hepatitis in Rhesus Macaques: WE but Not ARM Strain Activates Hepatocytes and Induces Liver Regeneration. Arch. Virol. 2004, 149, 2319–2336. [Google Scholar] [CrossRef] [PubMed]
- Rodas, J.D.; Lukashevich, I.S.; Zapata, J.C.; Cairo, C.; Tikhonov, I.; Djavani, M.; Pauza, C.D.; Salvato, M.S. Mucosal Arenavirus Infection of Primates Can Protect Them from Lethal Hemorrhagic Fever. J. Med. Virol. 2004, 72, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.R.; Warner, B.M.; Audet, J.; Soule, G.; Siragam, V.; Sroga, P.; Griffin, B.D.; Leung, A.; Grolla, A.; Tierney, K. Differential Pathogenesis of Closely Related 2018 Nigerian Outbreak Clade III Lassa Virus Isolates. PLoS Pathog. 2021, 17, e1009966. [Google Scholar] [CrossRef] [PubMed]
- Yun, N.E.; Ronca, S.; Tamura, A.; Koma, T.; Seregin, A.V.; Dineley, K.T.; Miller, M.; Cook, R.; Shimizu, N.; Walker, A.G. Animal Model of Sensorineural Hearing Loss Associated with Lassa Virus Infection. J. Virol. 2016, 90, 2920–2927. [Google Scholar] [CrossRef]
- Glushakova, S.E.; Lukashevich, I.S. Early Events in Arenavirus Replication Are Sensitive to Lysosomotropic Compounds. Arch. Virol. 1989, 104, 157–161. [Google Scholar] [CrossRef]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.-Y.; Kunz, S. Old World Arenaviruses Enter the Host Cell via the Multivesicular Body and Depend on the Endosomal Sorting Complex Required for Transport. PLoS Pathog. 2011, 7, e1002232. [Google Scholar] [CrossRef]
- Quirin, K.; Eschli, B.; Scheu, I.; Poort, L.; Kartenbeck, J.; Helenius, A. Lymphocytic Choriomeningitis Virus Uses a Novel Endocytic Pathway for Infectious Entry via Late Endosomes. Virology 2008, 378, 21–33. [Google Scholar] [CrossRef]
- Rojek, J.M.; Sanchez, A.B.; Nguyen, N.T.; de la Torre, J.-C.; Kunz, S. Different Mechanisms of Cell Entry by Human-Pathogenic Old World and New World Arenaviruses. J. Virol. 2008, 82, 7677–7687. [Google Scholar] [CrossRef]
- Iwasaki, M.; Ngo, N.; de la Torre, J.C. Sodium Hydrogen Exchangers Contribute to Arenavirus Cell Entry. J. Virol. 2014, 88, 643–654. [Google Scholar] [CrossRef]
- Oppliger, J.; Torriani, G.; Herrador, A.; Kunz, S. Lassa Virus Cell Entry via Dystroglycan Involves an Unusual Pathway of Macropinocytosis. J. Virol. 2016, 90, 6412–6429. [Google Scholar] [CrossRef]
- Cohen-Dvashi, H.; Cohen, N.; Israeli, H.; Diskin, R. Molecular Mechanism for LAMP1 Recognition by Lassa Virus. J. Virol. 2015, 89, 7584–7592. [Google Scholar] [CrossRef]
- Jae, L.T.; Raaben, M.; Herbert, A.S.; Kuehne, A.I.; Wirchnianski, A.S.; Soh, T.K.; Stubbs, S.H.; Janssen, H.; Damme, M.; Saftig, P. Lassa Virus Entry Requires a Trigger-Induced Receptor Switch. Science 2014, 344, 1506–1510. [Google Scholar] [CrossRef]
- Brouillette, R.B.; Phillips, E.K.; Patel, R.; Mahauad-Fernandez, W.; Moller-Tank, S.; Rogers, K.J.; Dillard, J.A.; Cooney, A.L.; Martinez-Sobrido, L.; Okeoma, C. TIM-1 Mediates Dystroglycan-Independent Entry of Lassa Virus. J. Virol. 2018, 93, e02185-18. [Google Scholar] [CrossRef]
- Fedeli, C.; Torriani, G.; Galan-Navarro, C.; Moraz, M.-L.; Moreno, H.; Gerold, G.; Kunz, S. Axl Can Serve as Entry Factor for Lassa Virus Depending on the Functional Glycosylation of Dystroglycan. J. Virol. 2018, 92, e01613-17. [Google Scholar] [CrossRef]
- Goncalves, A.-R.; Moraz, M.-L.; Pasquato, A.; Helenius, A.; Lozach, P.-Y.; Kunz, S. Role of DC-SIGN in Lassa Virus Entry into Human Dendritic Cells. J. Virol. 2013, 87, 11504–11515. [Google Scholar] [CrossRef]
- Shimojima, M.; Kawaoka, Y. Cell Surface Molecules Involved in Infection Mediated by Lymphocytic Choriomeningitis Virus Glycoprotein. J. Vet. Med. Sci. 2012, 74, 1363–1366. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Ströher, U.; Ebihara, H.; Feldmann, H.; Kawaoka, Y. Identification of Cell Surface Molecules Involved in Dystroglycan-Independent Lassa Virus Cell Entry. J. Virol. 2012, 86, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Fedeli, C.; Moreno, H.; Kunz, S. The Role of Receptor Tyrosine Kinases in Lassa Virus Cell Entry. Viruses 2020, 12, 857. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S.; Paessler, S.; de la Torre, J.C. Lassa Virus Diversity and Feasibility for Universal Prophylactic Vaccine. F1000Research 2019, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.; Mercer, J. Viral Apoptotic Mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Zagórska, A.; Lew, E.D.; Shrestha, B.; Rothlin, C.V.; Naughton, J.; Diamond, M.S.; Lemke, G.; Young, J.A. Enveloped Viruses Disable Innate Immune Responses in Dendritic Cells by Direct Activation of TAM Receptors. Cell Host Microbe 2013, 14, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Moller-Tank, S.; Maury, W. Phosphatidylserine Receptors: Enhancers of Enveloped Virus Entry and Infection. Virology 2014, 468, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Briese, T.; Lipkin, W.I. Z Proteins of New World Arenaviruses Bind RIG-I and Interfere with Type I Interferon Induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef]
- Jiang, X.; Huang, Q.; Wang, W.; Dong, H.; Ly, H.; Liang, Y.; Dong, C. Structures of Arenaviral Nucleoproteins with Triphosphate dsRNA Reveal a Unique Mechanism of Immune Suppression. J. Biol. Chem. 2013, 288, 16949–16959. [Google Scholar] [CrossRef]
- Pythoud, C.; Rodrigo, W.S.I.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus Nucleoprotein Targets Interferon Regulatory Factor-Activating Kinase IKKε. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef]
- Xing, J.; Ly, H.; Liang, Y. The Z Proteins of Pathogenic but Not Nonpathogenic Arenaviruses Inhibit RIG-I-like Receptor-Dependent Interferon Production. J. Virol. 2015, 89, 2944–2955. [Google Scholar] [CrossRef]
- Borrow, P.; Martínez-Sobrido, L.; De la Torre, J.C. Inhibition of the Type I Interferon Antiviral Response during Arenavirus Infection. Viruses 2010, 2, 2443–2480. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Lymphocyte Choriomeningitis Virus Plays Hide-and-Seek with Type 1 Interferon. Cell Host Microbe 2012, 11, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.W.; Carrion, R., Jr.; Nunneley, J.; Medvedev, A.E.; Salvato, M.S.; Lukashevich, I.S. Pathogenic Old World Arenaviruses Inhibit TLR2/Mal-Dependent Proinflammatory Cytokines in Vitro. J. Virol. 2012, 86, 7216–7226. [Google Scholar] [CrossRef] [PubMed]
- Carrion, R., Jr.; Brasky, K.; Mansfield, K.; Johnson, C.; Gonzales, M.; Ticer, A.; Lukashevich, I.; Tardif, S.; Patterson, J. Lassa Virus Infection in Experimentally Infected Marmosets: Liver Pathology and Immunophenotypic Alterations in Target Tissues. J. Virol. 2007, 81, 6482–6490. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Maryankova, R.; Vladyko, A.S.; Nashkevich, N.; Koleda, S.; Djavani, M.; Horejsh, D.; Voitenok, N.N.; Salvato, M.S. Lassa and Mopeia Virus Replication in Human Monocytes/Macrophages and in Endothelial Cells: Different Effects on IL-8 and TNF-α Gene Expression. J. Med. Virol. 1999, 59, 552–560. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Carrion, R., Jr.; Salvato, M.S.; Mansfield, K.; Brasky, K.; Zapata, J.; Cairo, C.; Goicochea, M.; Hoosien, G.E.; Ticer, A. Safety, Immunogenicity, and Efficacy of the ML29 Reassortant Vaccine for Lassa Fever in Small Non-Human Primates. Vaccine 2008, 26, 5246–5254. [Google Scholar] [CrossRef] [PubMed]
- Warner, N.L.; Jokinen, J.D.; Beier, J.I.; Sokoloski, K.J.; Lukashevich, I.S. Mammarenaviral Infection Is Dependent on Directional Exposure to and Release from Polarized Intestinal Epithelia. Viruses 2018, 10, 75. [Google Scholar] [CrossRef] [PubMed]
- Cosset, F.-L.; Marianneau, P.; Verney, G.; Gallais, F.; Tordo, N.; Pécheur, E.-I.; ter Meulen, J.; Deubel, V.; Bartosch, B. Characterization of Lassa Virus Cell Entry and Neutralization with Lassa Virus Pseudoparticles. J. Virol. 2009, 83, 3228–3237. [Google Scholar] [CrossRef]
- Rojek, J.M.; Perez, M.; Kunz, S. Cellular Entry of Lymphocytic Choriomeningitis Virus. J. Virol. 2008, 82, 1505–1517. [Google Scholar] [CrossRef]
- Danthi, P.; Chow, M. Cholesterol Removal by Methyl-β-Cyclodextrin Inhibits Poliovirus Entry. J. Virol. 2004, 78, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.; O’NEILL, C.A.; Padfield, P.J. Depletion of Caco-2 Cell Cholesterol Disrupts Barrier Function by Altering the Detergent Solubility and Distribution of Specific Tight-Junction Proteins. Biochem. J. 2005, 387, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Jokinen, J.; Tretyakova, I.; Pushko, P.; Lukashevich, I.S. Alphavirus Vector-Based Replicon Particles Expressing Multivalent Cross-Protective Lassa Virus Glycoproteins. Vaccine 2018, 36, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Idrees, S.; Chen, H.; Panth, N.; Paudel, K.R.; Hansbro, P.M. Exploring Viral–Host Protein Interactions as Antiviral Therapies: A Computational Perspective. Microorganisms 2024, 12, 630. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Minder, P.; Cai, Y.; Kuhn, J.H.; Yates III, J.R.; Torbett, B.E.; de la Torre, J.C. Interactome Analysis of the Lymphocytic Choriomeningitis Virus Nucleoprotein in Infected Cells Reveals ATPase Na+/K+ Transporting Subunit Alpha 1 and Prohibitin as Host-Cell Factors Involved in the Life Cycle of Mammarenaviruses. PLoS Pathog. 2018, 14, e1006892. [Google Scholar] [CrossRef]
- Hulseberg, C.E.; Fénéant, L.; Szymańska, K.M.; White, J.M. Lamp1 Increases the Efficiency of Lassa Virus Infection by Promoting Fusion in Less Acidic Endosomal Compartments. MBio 2018, 9, e01818-17. [Google Scholar] [CrossRef]
- Spearman, P. Viral Interactions with Host Cell Rab GTPases. Small GTPases 2018, 9, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-Signaling Networks: An Integration of Adaptor Molecules, Kinases, and Cross-Talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef]
- Kawagoe, T.; Sato, S.; Matsushita, K.; Kato, H.; Matsui, K.; Kumagai, Y.; Saitoh, T.; Kawai, T.; Takeuchi, O.; Akira, S. Sequential Control of Toll-like Receptor–Dependent Responses by IRAK1 and IRAK2. Nat. Immunol. 2008, 9, 684–691. [Google Scholar] [CrossRef]
- McCormick, J.B.; Walker, D.H.; King, I.J.; Webb, P.A.; Elliott, L.H.; Whitfield, S.G.; Johnson, K.M. Lassa Virus Hepatitis: A Study of Fatal Lassa Fever in Humans. Am. J. Trop. Med. Hyg. 1986, 35, 401–407. [Google Scholar] [CrossRef]
- Kunz, S.; Rojek, J.M.; Kanagawa, M.; Spiropoulou, C.F.; Barresi, R.; Campbell, K.P.; Oldstone, M.B. Posttranslational Modification of α-Dystroglycan, the Cellular Receptor for Arenaviruses, by the Glycosyltransferase LARGE Is Critical for Virus Binding. J. Virol. 2005, 79, 14282–14296. [Google Scholar] [CrossRef]
- Kunz, S.; Rojek, J.M.; Perez, M.; Spiropoulou, C.F.; Oldstone, M.B. Characterization of the Interaction of Lassa Fever Virus with Its Cellular Receptor α-Dystroglycan. J. Virol. 2005, 79, 5979–5987. [Google Scholar] [CrossRef]
- Hastie, K.M.; Igonet, S.; Sullivan, B.M.; Legrand, P.; Zandonatti, M.A.; Robinson, J.E.; Garry, R.F.; Rey, F.A.; Oldstone, M.B.; Saphire, E.O. Crystal Structure of the Prefusion Surface Glycoprotein of the Prototypic Arenavirus LCMV. Nat. Struct. Mol. Biol. 2016, 23, 513–521. [Google Scholar] [CrossRef]
- Kunz, S.; Sevilla, N.; McGavern, D.B.; Campbell, K.P.; Oldstone, M.B. Molecular Analysis of the Interaction of LCMV with Its Cellular Receptor α-Dystroglycan. J. Cell Biol. 2001, 155, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, N.; Kunz, S.; McGavern, D.; Oldstone, M.B.A. Infection of Dendritic Cells by Lymphocytic Choriomeningitis Virus. Dendritic Cells Virus Infect. 2003, 276, 125–144. [Google Scholar]
- Smelt, S.C.; Borrow, P.; Kunz, S.; Cao, W.; Tishon, A.; Lewicki, H.; Campbell, K.P.; Oldstone, M.B. Differences in Affinity of Binding of Lymphocytic Choriomeningitis Virus Strains to the Cellular Receptor α-Dystroglycan Correlate with Viral Tropism and Disease Kinetics. J. Virol. 2001, 75, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.T.; Oldstone, M.B. Infected CD8α− Dendritic Cells Are the Predominant Source of IL-10 during Establishment of Persistent Viral Infection. Proc. Natl. Acad. Sci. USA 2012, 109, 14116–14121. [Google Scholar] [CrossRef]
- Borrow, P.; Evans, C.F.; Oldstone, M.B. Virus-Induced Immunosuppression: Immune System-Mediated Destruction of Virus-Infected Dendritic Cells Results in Generalized Immune Suppression. J. Virol. 1995, 69, 1059–1070. [Google Scholar] [CrossRef]
- Sullivan, B.M.; Emonet, S.F.; Welch, M.J.; Lee, A.M.; Campbell, K.P.; de la Torre, J.C.; Oldstone, M.B. Point Mutation in the Glycoprotein of Lymphocytic Choriomeningitis Virus Is Necessary for Receptor Binding, Dendritic Cell Infection, and Long-Term Persistence. Proc. Natl. Acad. Sci. USA 2011, 108, 2969–2974. [Google Scholar] [CrossRef]
- Xu, H.C.; Pandey, P.; Ward, H.; Gorzkiewicz, M.; Abromavičiūtė, D.; Tinz, C.; Müller, L.; Meyer, C.; Pandyra, A.A.; Yavas, A. High-Affinity–Mediated Viral Entry Triggers Innate Affinity Escape Resulting in Type I IFN Resistance and Impaired T Cell Immunity. J. Immunol. 2024, 212, 1457–1466. [Google Scholar] [CrossRef]
- Namineni, S.; O’Connor, T.; Faure-Dupuy, S.; Johansen, P.; Riedl, T.; Liu, K.; Xu, H.; Singh, I.; Shinde, P.; Li, F. A Dual Role for Hepatocyte-Intrinsic Canonical NF-κB Signaling in Virus Control. J. Hepatol. 2020, 72, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Liu, X.; Brisse, M.; Ly, H.; Liang, Y. Effect of Strain Variations on Lassa Virus Z Protein-Mediated Human RIG-I Inhibition. Viruses 2020, 12, 907. [Google Scholar] [CrossRef] [PubMed]
- Shah, W.A.; Peng, H.; Carbonetto, S. Role of Non-Raft Cholesterol in Lymphocytic Choriomeningitis Virus Infection via α-Dystroglycan. J. Gen. Virol. 2006, 87, 673–678. [Google Scholar] [CrossRef]
- Cuevas, C.D.; Ross, S.R. Toll-like Receptor 2-Mediated Innate Immune Responses against Junín Virus in Mice Lead to Antiviral Adaptive Immune Responses during Systemic Infection and Do Not Affect Viral Replication in the Brain. J. Virol. 2014, 88, 7703–7714. [Google Scholar] [CrossRef]
- Cuevas, C.D.; Lavanya, M.; Wang, E.; Ross, S.R. Junin Virus Infects Mouse Cells and Induces Innate Immune Responses. J. Virol. 2011, 85, 11058–11068. [Google Scholar] [CrossRef]
- Zhou, S.; Cerny, A.M.; Bowen, G.; Chan, M.; Knipe, D.M.; Kurt-Jones, E.A.; Finberg, R.W. Discovery of a Novel TLR2 Signaling Inhibitor with Anti-Viral Activity. Antivir. Res. 2010, 87, 295–306. [Google Scholar] [CrossRef]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic Choriomeningitis Virus (LCMV) Infection of CNS Glial Cells Results in TLR2-MyD88/Mal-Dependent Inflammatory Responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef]
- Zhou, S.; Kurt-Jones, E.A.; Mandell, L.; Cerny, A.; Chan, M.; Golenbock, D.T.; Finberg, R.W. MyD88 Is Critical for the Development of Innate and Adaptive Immunity during Acute Lymphocytic Choriomeningitis Virus Infection. Eur. J. Immunol. 2005, 35, 822–830. [Google Scholar] [CrossRef]
- Helenius, A. Virus Entry: Looking Back and Moving Forward. J. Mol. Biol. 2018, 430, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Bakkers, M.J.; Moon-Walker, A.; Herlo, R.; Brusic, V.; Stubbs, S.H.; Hastie, K.M.; Saphire, E.O.; Kirchhausen, T.L.; Whelan, S.P. CD164 Is a Host Factor for Lymphocytic Choriomeningitis Virus Entry. Proc. Natl. Acad. Sci. USA 2022, 119, e2119676119. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.L.; Barton, G.M. Trafficking of Endosomal Toll-like Receptors. Trends Cell Biol. 2014, 24, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- Stachura, P.; Stencel, O.; Lu, Z.; Borkhardt, A.; Pandyra, A.A. Arenaviruses: Old Viruses Present New Solutions for Cancer Therapy. Front. Immunol. 2023, 14, 1110522. [Google Scholar] [CrossRef]
- Kalkavan, H.; Sharma, P.; Kasper, S.; Helfrich, I.; Pandyra, A.A.; Gassa, A.; Virchow, I.; Flatz, L.; Brandenburg, T.; Namineni, S. Spatiotemporally Restricted Arenavirus Replication Induces Immune Surveillance and Type I Interferon-Dependent Tumour Regression. Nat. Commun. 2017, 8, 14447. [Google Scholar] [CrossRef]
- Bhat, H.; Zaun, G.; Hamdan, T.A.; Lang, J.; Adomati, T.; Schmitz, R.; Friedrich, S.-K.; Bergerhausen, M.; Cham, L.B.; Li, F. Arenavirus Induced CCL5 Expression Causes NK Cell-Mediated Melanoma Regression. Front. Immunol. 2020, 11, 1849. [Google Scholar] [CrossRef] [PubMed]
- Baharom, F.; Thomas, O.S.; Lepzien, R.; Mellman, I.; Chalouni, C.; Smed-Sörensen, A. Visualization of Early Influenza A Virus Trafficking in Human Dendritic Cells Using STED Microscopy. PLoS ONE 2017, 12, e0177920. [Google Scholar] [CrossRef] [PubMed]
- King, B.R.; Samacoits, A.; Eisenhauer, P.L.; Ziegler, C.M.; Bruce, E.A.; Zenklusen, D.; Zimmer, C.; Mueller, F.; Botten, J. Visualization of Arenavirus RNA Species in Individual Cells by Single-Molecule Fluorescence in Situ Hybridization Suggests a Model of Cyclical Infection and Clearance during Persistence. J. Virol. 2018, 92, e02241-17. [Google Scholar] [CrossRef]
- Ziegler, C.M.; Bruce, E.A.; Kelly, J.A.; King, B.R.; Botten, J.W. The Use of Novel Epitope-Tagged Arenaviruses Reveals That Rab5c-Positive Endosomal Membranes Are Targeted by the LCMV Matrix Protein. J. Gen. Virol. 2018, 99, 187. [Google Scholar] [CrossRef]
- Hongu, T.; Kanaho, Y. Activation Machinery of the Small GTPase Arf6. Adv. Biol. Regul. 2014, 54, 59–66. [Google Scholar] [CrossRef]
- Blasius, A.L.; Beutler, B. Intracellular Toll-like Receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, D.M.; Khakhum, N.; Wang, M.; Warner, N.L.; Jokinen, J.D.; Comer, J.E.; Lukashevich, I.S. Pathogenic and Apathogenic Strains of Lymphocytic Choriomeningitis Virus Have Distinct Entry and Innate Immune Activation Pathways. Viruses 2024, 16, 635. https://doi.org/10.3390/v16040635
Johnson DM, Khakhum N, Wang M, Warner NL, Jokinen JD, Comer JE, Lukashevich IS. Pathogenic and Apathogenic Strains of Lymphocytic Choriomeningitis Virus Have Distinct Entry and Innate Immune Activation Pathways. Viruses. 2024; 16(4):635. https://doi.org/10.3390/v16040635
Chicago/Turabian StyleJohnson, Dylan M., Nittaya Khakhum, Min Wang, Nikole L. Warner, Jenny D. Jokinen, Jason E. Comer, and Igor S. Lukashevich. 2024. "Pathogenic and Apathogenic Strains of Lymphocytic Choriomeningitis Virus Have Distinct Entry and Innate Immune Activation Pathways" Viruses 16, no. 4: 635. https://doi.org/10.3390/v16040635