
Autophagy Deregulation in HIV-1-Infected Cells Increases Extracellular Vesicle Release and Contributes to TLR3 Activation
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Primary Macrophages
2.3. Preparation of Whole Cell Extracts
2.4. Enrichment of EVs with Nanotrap Particles
2.5. Western Blot Analysis
2.6. Ultracentrifugation for EV Isolation
2.7. EV Concentration Analysis
2.8. Cell Viability Assay
2.9. In Vitro Kinase Assay
2.10. Construction of Two-Dimensional and Three-Dimensional TAR RNA Structures
2.11. TAR-TLR3 Docking Analysis
2.12. Densitometry Analysis
2.13. Real Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.14. Statistical Analysis
3. Results
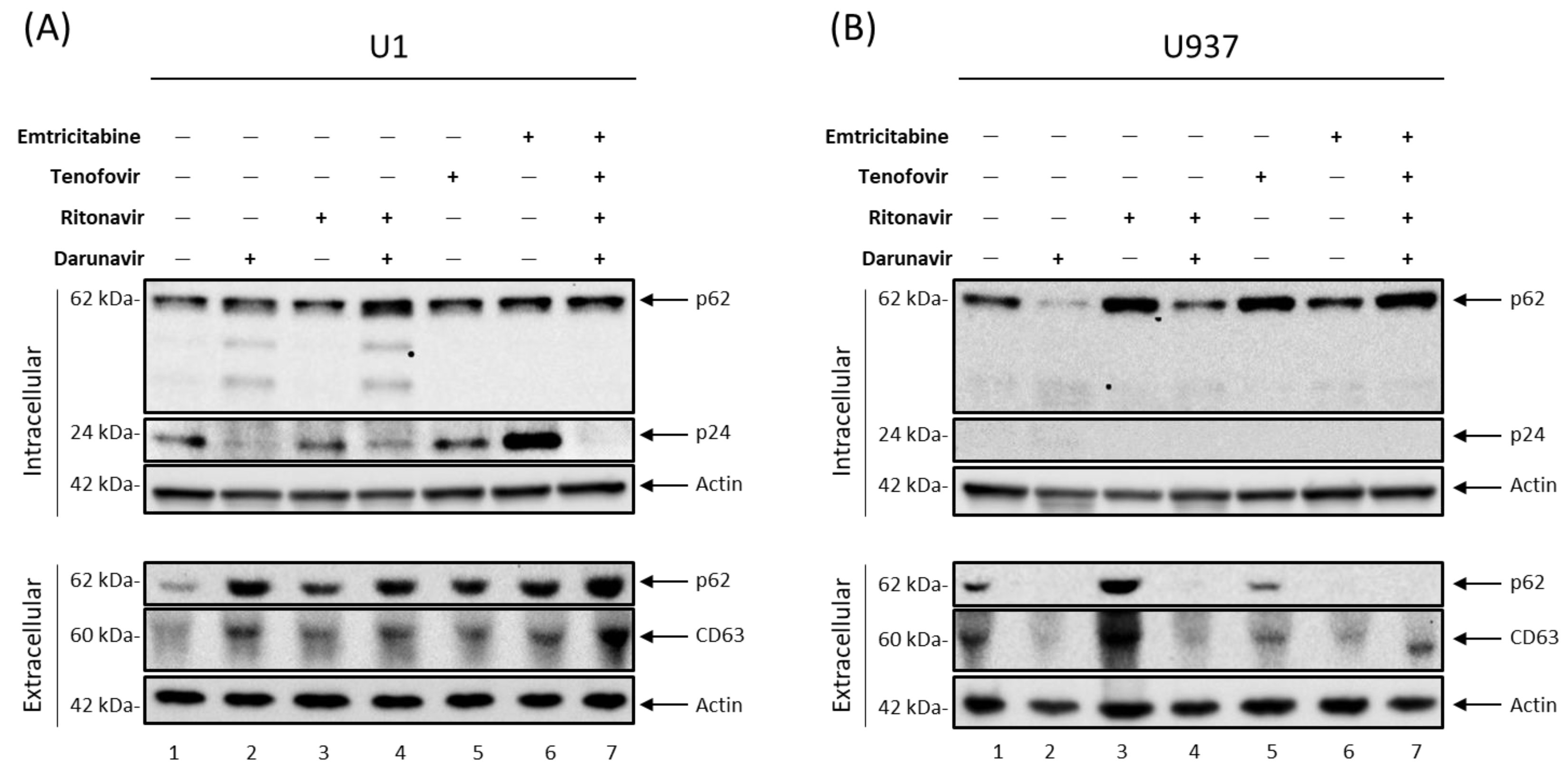
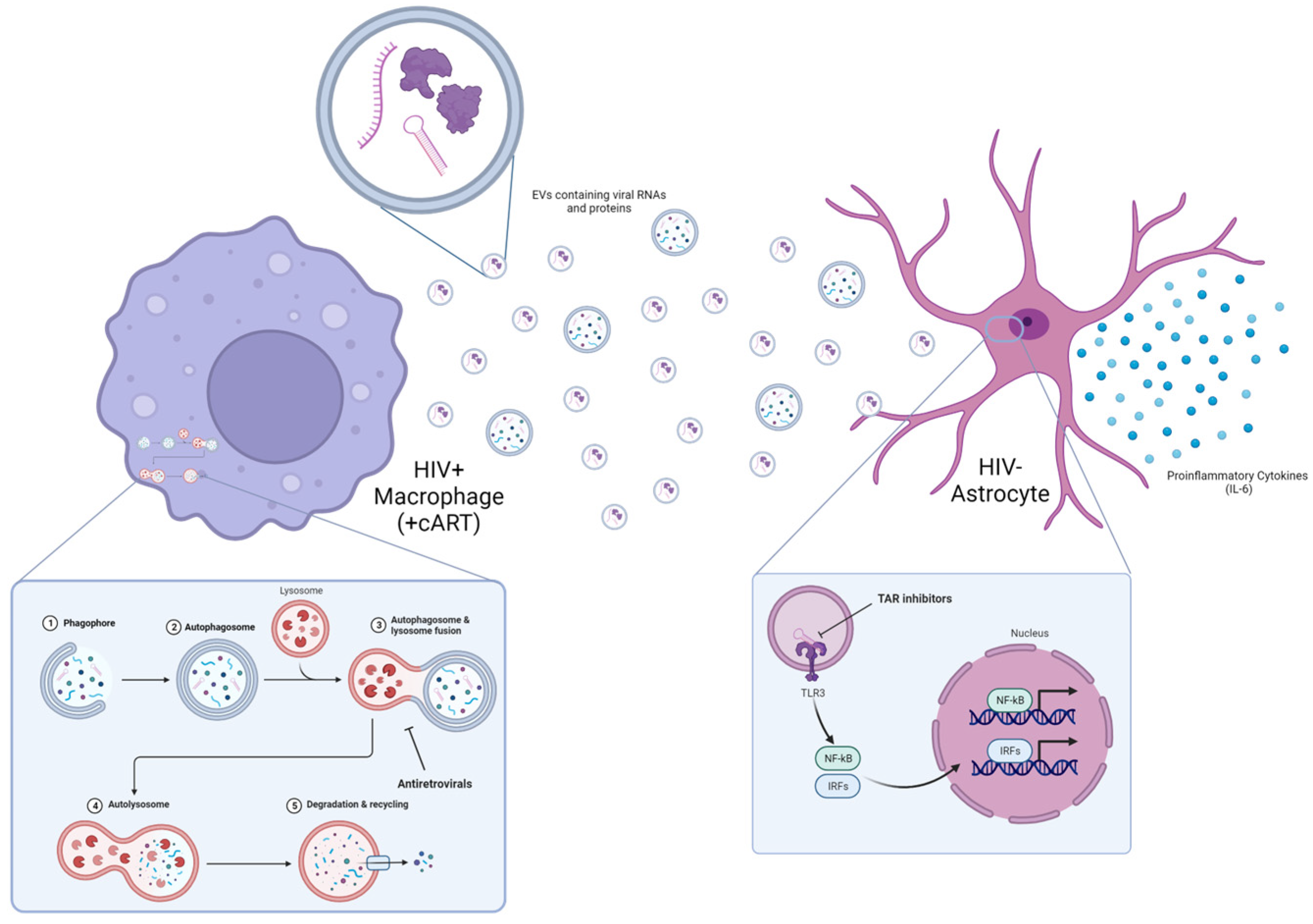
3.1. Antiretroviral Drugs Alter EV Release from HIV-1-Infected Monocytes and Macrophages
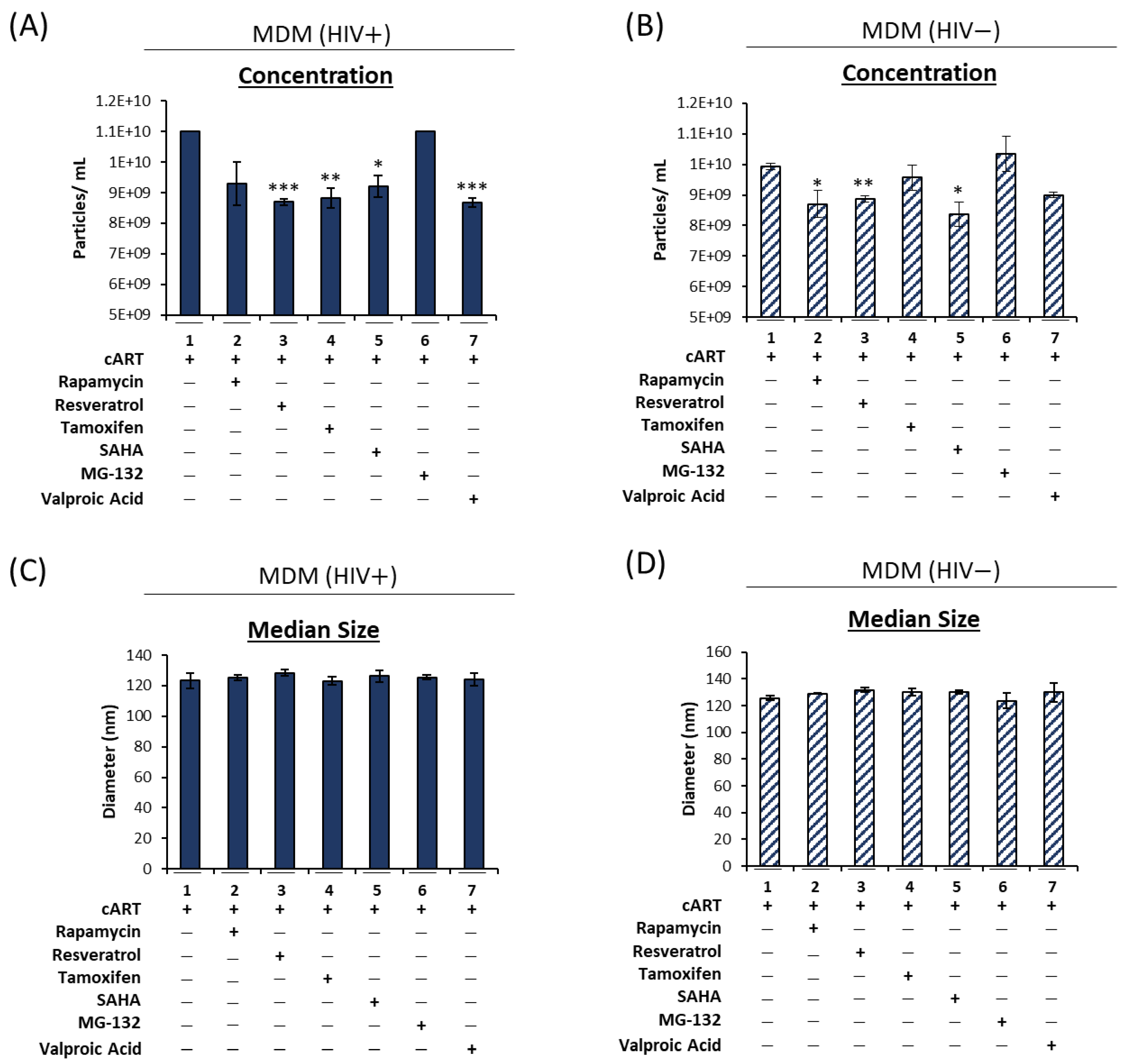
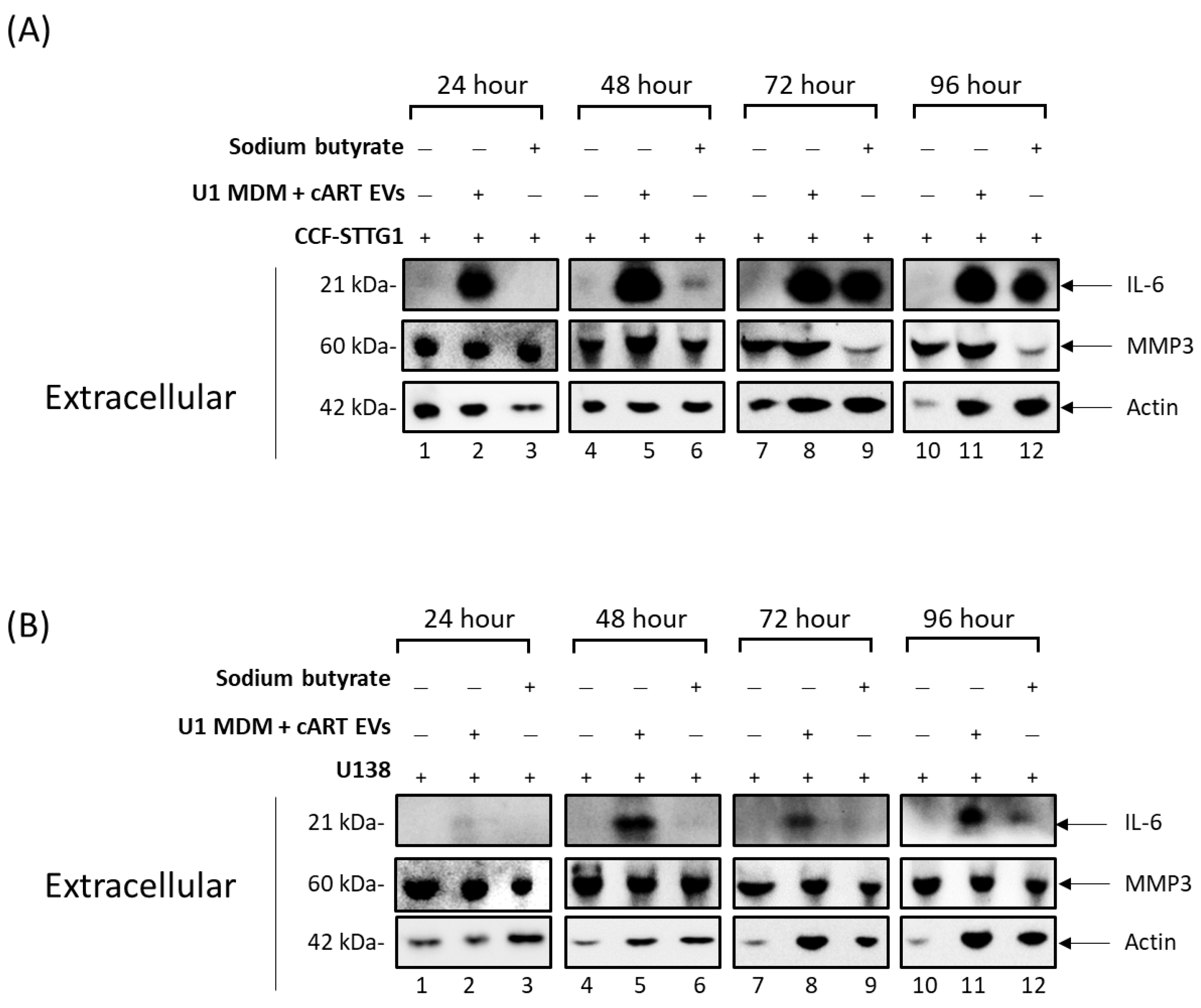
3.2. cART-Mediated Changes in Autophagy Contribute to HIV-1 Pathogenesis
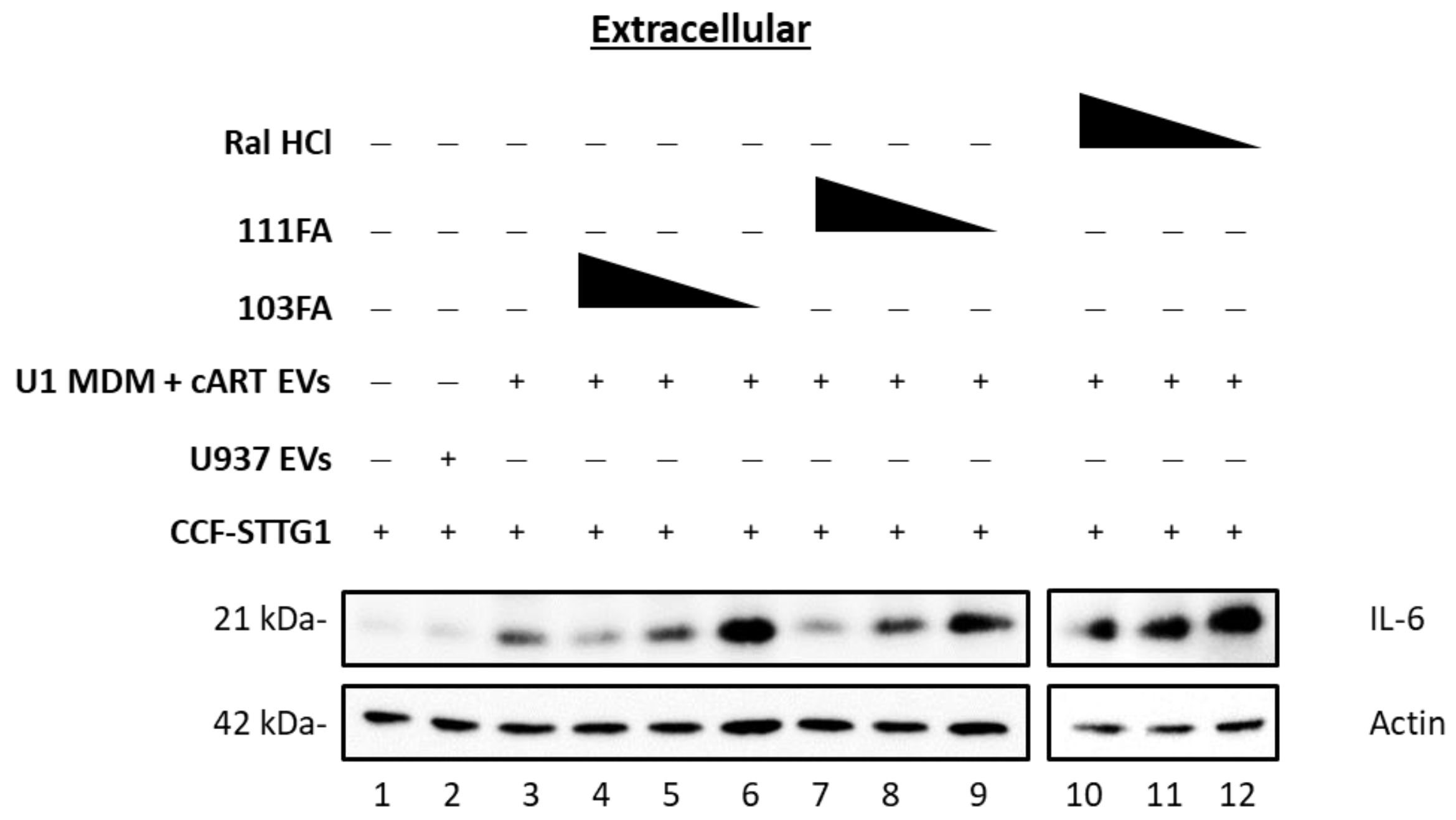
3.3. Small-Molecule TAR-Binding Compounds Inhibit TLR3 Activation and Mitigate EV-Mediated Effects on Astrocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS. AIDS by the Numbers, AIDS Is Not Over, But It Can Be; Joint United Nations Programme on HIV/AIDS (UNAIDS): Geneva, Switzerland, 2016; p. 28. [Google Scholar]
- Simonetti, F.R.; Kearney, M.F. Review: Influence of ART on HIV Genetics. Curr. Opin. HIV AIDS 2015, 10, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. Department of Health and Human Services. Available online: https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv (accessed on 6 January 2019).
- Ellero, J.; Lubomski, M.; Brew, B. Interventions for Neurocognitive Dysfunction. Curr. HIV/AIDS Rep. 2017, 14, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Eggers, C.; Arendt, G.; Hahn, K.; Husstedt, I.W.; Maschke, M.; Neuen-Jacob, E.; Obermann, M.; Rosenkranz, T.; Schielke, E.; Straube, E. HIV-1-Associated Neurocognitive Disorder: Epidemiology, Pathogenesis, Diagnosis, and Treatment. J. Neurol. 2017, 264, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Rourke, S. Asymptomatic Neurocognitive Impairment (ANI) Is Associated with Progression to Symptomatic HIV-Associated Neurocognitive Disorders (HAND) in People with HIV: Results from The Ontario HIV Treatment Network (OHTN) Cohort Study. Available online: http://www.natap.org/2015/IAS/IAS_90.htm (accessed on 6 January 2019).
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-Associated Neurocognitive Disorders before and during the Era of Combination Antiretroviral Therapy: Differences in Rates, Nature, and Predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- DeMarino, C.; Pleet, M.L.; Cowen, M.; Barclay, R.A.; Akpamagbo, Y.; Erickson, J.; Ndembe, N.; Charurat, M.; Jumare, J.; Bwala, S.; et al. Antiretroviral Drugs Alter the Content of Extracellular Vesicles from HIV-1-Infected Cells. Sci. Rep. 2018, 8, 7653. [Google Scholar] [CrossRef] [PubMed]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.-H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-Based Measures of Viral Persistence Are Associated with Immune Activation and Programmed Cell Death Protein 1 (PD-1)-Expressing CD4+ T Cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Hladnik, A.; Ferdin, J.; Goričar, K.; Deeks, G.S.; Peterlin, M.B.; Plemenitaš, A.; Vita, D.; Metka, L. Trans-Activation Response Element RNA Is Detectable in the Plasma of a Subset of Aviremic HIV-1-Infected Patients. Acta Chim. Slov. 2017, 64, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Veenstra, M.; Gaskill, P.J.; Morgello, S.; Calderon, T.M.; Berman, J.W. Monocytes Mediate HIV Neuropathogenesis: Mechanisms That Contribute to HIV Associated Neurocognitive Disorders. Curr. HIV Res. 2014, 12, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Nath, A. Human Immunodeficiency Virus (HIV) Proteins in Neuropathogenesis of HIV Dementia. J. Infect. Dis. 2002, 186, S193–S198. [Google Scholar] [CrossRef]
- Otaguiri, K.K.; Dos Santos, D.F.; Slavov, S.N.; Depieri, L.V.; Palma, P.V.B.; Meirelles, F.V.; Covas, D.T.; da Silveira, J.C.; Kashima, S. TAX-mRNA-Carrying Exosomes from Human T Cell Lymphotropic Virus Type 1-Infected Cells Can Induce Interferon-Gamma Production In Vitro. AIDS Res. Hum. Retroviruses 2018, 34, 1075–1082. [Google Scholar] [CrossRef]
- Anderson, M.R.; Pleet, M.L.; Enose-Akahata, Y.; Erickson, J.; Monaco, M.C.; Akpamagbo, Y.; Velluci, A.; Tanaka, Y.; Azodi, S.; Lepene, B.; et al. Viral Antigens Detectable in CSF Exosomes from Patients with Retrovirus Associated Neurologic Disease: Functional Role of Exosomes. Clin. Transl. Med. 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G.; Gunawardena, H.P.; Dekroon, R.M.; Heaton, P.R.; Edwards, R.H.; Ozgur, S.; Griffith, J.D.; Damania, B.; Raab-Traub, N. Modulation of B-Cell Exosome Proteins by Gamma Herpesvirus Infection. Proc. Natl. Acad. Sci. USA 2013, 110, E2925–E2933. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G.; Shair, K.H.Y.; Marquitz, A.R.; Kung, C.-P.; Edwards, R.H.; Raab-Traub, N. Human Tumor Virus Utilizes Exosomes for Intercellular Communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Meyering, S.S.; Zadeh, M.A.; Saifuddin, M.; Hakami, R.M.; Kashanchi, F. Exosomes and Their Role in CNS Viral Infections. J. Neurovirol. 2014, 20, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes Derived from HIV-1-Infected Cells Contain Trans-Activation Response Element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Asad Zadeh, M.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-Infected Cells Stimulate Production of Pro-Inflammatory Cytokines through Trans-Activating Response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Barclay, R.A.; Schwab, A.; DeMarino, C.; Akpamagbo, Y.; Lepene, B.; Kassaye, S.; Iordanskiy, S.; Kashanchi, F. Exosomes from Uninfected Cells Activate Transcription of Latent HIV-1. J. Biol. Chem. 2017, 292, 11682–11701. [Google Scholar] [CrossRef] [PubMed]
- Akpamagbo, Y.A.; DeMarino, C.; Pleet, M.L.; Schwab, A.; Rodriguez, M.; Barclay, R.A.; Sampey, G.; Iordanskiy, S.; El-Hage, N.; Kashanchi, F. HIV-1 Transcription Inhibitors Increase the Synthesis of Viral Non-Coding RNA That Contribute to Latency. Curr. Pharm. Des. 2017, 23, 4133–4144. [Google Scholar] [CrossRef] [PubMed]
- Pleet, M.L.; Branscome, H.; DeMarino, C.; Pinto, D.O.; Zadeh, M.A.; Rodriguez, M.; Sariyer, I.K.; El-Hage, N.; Kashanchi, F. Autophagy, EVs, and Infections: A Perfect Question for a Perfect Time. Front. Cell. Infect. Microbiol. 2018, 8, 362. [Google Scholar] [CrossRef]
- Santos-Llamas, A.; Monte, M.J.; Marin, J.J.G.; Perez, M.J. Dysregulation of Autophagy in Rat Liver with Mitochondrial DNA Depletion Induced by the Nucleoside Analogue Zidovudine. Arch. Toxicol. 2018, 92, 2109–2118. [Google Scholar] [CrossRef]
- Lin, H.; Stankov, M.V.; Hegermann, J.; Budida, R.; Panayotova-Dimitrova, D.; Schmidt, R.E.; Behrens, G.M.N. Zidovudine-Mediated Autophagy Inhibition Enhances Mitochondrial Toxicity in Muscle Cells. Antimicrob. Agents Chemother. 2019, 63, e01443-18. [Google Scholar] [CrossRef] [PubMed]
- Stankov, M.V.; Panayotova-Dimitrova, D.; Leverkus, M.; Schmidt, R.E.; Behrens, G.M. Thymidine Analogues Suppress Autophagy and Adipogenesis in Cultured Adipocytes. Antimicrob. Agents Chemother. 2013, 57, 543–551. [Google Scholar] [CrossRef]
- Cheney, L.; Calderon, T.; Cuervo, A.M.; Berman, J.W. Aberrant Autophagy in Macrophages and Astrocytes after HIV Nef or Antiretroviral Treatment: Contribution to the Pathogenesis of HIV-Associated Neurocognitive Disorders. Open Forum Infect. Dis. 2017, 4, S208. [Google Scholar] [CrossRef]
- Midde, N.; Sinha, N.; Lukka, P.; Meibohm, B.; Kumar, S. Alterations in Cellular Pharmacokinetics and Pharmacodynamics of Elvitegravir in Response to Ethanol Exposure in HIV-1 Infected Monocytic (U1) Cells. PLoS ONE 2017, 12, e0172628. [Google Scholar] [CrossRef] [PubMed]
- Graziano, F.; Elia, C.; Laudanna, C.; Poli, G.; Alfano, M. Urokinase Plasminogen Activator Inhibits HIV Virion Release from Macrophage-Differentiated Chronically Infected Cells via Activation of RhoA and PKCε. PLoS ONE 2011, 6, e23674. [Google Scholar] [CrossRef]
- Abulwerdi, F.A.; Shortridge, M.D.; Sztuba-Solinska, J.; Wilson, R.; Le Grice, S.F.J.; Varani, G.; Schneekloth, J.S. Development of Small Molecules with a Non-Canonical Binding Mode to HIV-1 Trans Activation Response (TAR) RNA. J. Med. Chem. 2016, 59, 11148–11160. [Google Scholar] [CrossRef]
- Zuker, M.; Stiegler, P. Optimal Computer Folding of Large RNA Sequences Using Thermodynamics and Auxiliary Information. Nucleic Acids Res. 1981, 9, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D Structure Composition for Large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef]
- Pinto, D.O.; Scott, T.A.; DeMarino, C.; Pleet, M.L.; Vo, T.T.; Saifuddin, M.; Kovalskyy, D.; Erickson, J.; Cowen, M.; Barclay, R.A.; et al. Effect of Transcription Inhibition and Generation of Suppressive Viral Non-Coding RNAs. Retrovirology 2019, 16, 13. [Google Scholar] [CrossRef]
- Leonard, J.N.; Ghirlando, R.; Askins, J.; Bell, J.K.; Margulies, D.H.; Davies, D.R.; Segal, D.M. The TLR3 Signaling Complex Forms by Cooperative Receptor Dimerization. Proc. Natl. Acad. Sci. USA 2008, 105, 258–263. [Google Scholar] [CrossRef]
- Liu, L.; Botos, I.; Wang, Y.; Leonard, J.N.; Shiloach, J.; Segal, D.M.; Davies, D.R. Structural Basis of Toll-like Receptor 3 Signaling with Double-Stranded RNA. Science 2008, 320, 379–381. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Zhang, C.; Vasmatzis, G.; Cornette, J.L.; DeLisi, C. Determination of Atomic Desolvation Energies from the Structures of Crystallized Proteins. J. Mol. Biol. 1997, 267, 707–726. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Schrödinger, LLC. PyMOL-2.3.2_79-Win64-portable-py27.zip. Available online: https://pymol.org/(accessed on 29 June 2019).
- Maycotte, P.; Thorburn, A. Targeting Autophagy in Breast Cancer. World J. Clin. Oncol. 2014, 5, 224–240. [Google Scholar] [CrossRef]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of Autophagy in Histone Deacetylase Inhibitor-Induced Apoptotic and Nonapoptotic Cell Death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef]
- Madigan, M.; Munoz-Erazo, L.; Conway, R. Valproic Acid (VPA), a Class I and II Histone Deacetylase (HDAC) Inhibitor and Conjunctival Melanoma. Acta Ophthalmol. 2012, 90. [Google Scholar] [CrossRef]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.-G.; Park, H.; Ryu, S.H. Resveratrol Induces Autophagy by Directly Inhibiting mTOR through ATP Competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef]
- Qiao, C.-M.; Sun, M.-F.; Jia, X.-B.; Li, Y.; Zhang, B.-P.; Zhao, L.-P.; Shi, Y.; Zhou, Z.-L.; Zhu, Y.-L.; Cui, C.; et al. Sodium Butyrate Exacerbates Parkinson’s Disease by Aggravating Neuroinflammation and Colonic Inflammation in MPTP-Induced Mice Model. Neurochem. Res. 2020, 45, 2128–2142. [Google Scholar] [CrossRef]
- Trudler, D.; Farfara, D.; Frenkel, D. Toll-Like Receptors Expression and Signaling in Glia Cells in Neuro-Amyloidogenic Diseases: Towards Future Therapeutic Application. Available online: https://www.hindawi.com/journals/mi/2010/497987/ (accessed on 11 December 2020).
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad Expression of Toll-Like Receptors in the Human Central Nervous System. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef]
- El-Hage, N.; Podhaizer, E.M.; Sturgill, J.; Hauser, K.F. Toll-like Receptor Expression and Activation in Astroglia: Differential Regulation by HIV-1 Tat, Gp120, and Morphine. Immunol. Investig. 2011, 40, 498–522. [Google Scholar] [CrossRef]
- Lafon, M.; Megret, F.; Lafage, M.; Prehaud, C. The Innate Immune Facet of Brain. J. Mol. Neurosci. 2006, 29, 185–194. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.; Davies, D.R.; Segal, D.M. Dimerization of Toll-like Receptor 3 (TLR3) Is Required for Ligand Binding. J. Biol. Chem. 2010, 285, 36836–36841. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of Double-Stranded RNA and Activation of NF-kappaB by Toll-like Receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Bell, J.K.; Askins, J.; Hall, P.R.; Davies, D.R.; Segal, D.M. The dsRNA Binding Site of Human Toll-like Receptor 3. Proc. Natl. Acad. Sci. USA 2006, 103, 8792–8797. [Google Scholar] [CrossRef]
- Tatematsu, M.; Nishikawa, F.; Seya, T.; Matsumoto, M. Toll-like Receptor 3 Recognizes Incomplete Stem Structures in Single-Stranded Viral RNA. Nat. Commun. 2013, 4, 1833. [Google Scholar] [CrossRef]
- Botos, I.; Liu, L.; Wang, Y.; Segal, D.M.; Davies, D.R. The Toll-like Receptor 3:dsRNA Signaling Complex. Biochim. Biophys. Acta 2009, 1789, 667–674. [Google Scholar] [CrossRef]
- Kumar, A.M.; Borodowsky, I.; Fernandez, B.; Gonzalez, L.; Kumar, M. Human Immunodeficiency Virus Type 1 RNA Levels in Different Regions of Human Brain: Quantification Using Real-Time Reverse Transcriptase-Polymerase Chain Reaction. J. Neurovirol. 2007, 13, 210–224. [Google Scholar] [CrossRef]
- Glass, J.D.; Fedor, H.; Wesselingh, S.L.; McArthur, J.C. Immunocytochemical Quantitation of Human Immunodeficiency Virus in the Brain: Correlations with Dementia. Ann. Neurol. 1995, 38, 755–762. [Google Scholar] [CrossRef]
- Takahashi, K.; Wesselingh, S.L.; Griffin, D.E.; McArthur, J.C.; Johnson, R.T.; Glass, J.D. Localization of HIV-1 in Human Brain Using Polymerase Chain Reaction/in Situ Hybridization and Immunocytochemistry. Ann. Neurol. 1996, 39, 705–711. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s Disease. Cell. Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Sittler, A.; Muriel, M.-P.; Marinello, M.; Brice, A.; den Dunnen, W.; Alves, S. Deregulation of Autophagy in Postmortem Brains of Machado-Joseph Disease Patients. Neuropathology 2018, 38, 113–124. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s Disease β-Amyloid Peptides Are Released in Association with Exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-Produced Alpha-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef]
- Jy, W.; Minagar, A.; Jimenez, J.J.; Sheremata, W.A.; Mauro, L.M.; Horstman, L.L.; Bidot, C.; Ahn, Y.S. Endothelial Microparticles (EMP) Bind and Activate Monocytes: Elevated EMP-Monocyte Conjugates in Multiple Sclerosis. Front. Biosci. J. Virtual Libr. 2004, 9, 3137–3144. [Google Scholar] [CrossRef]
- Minagar, A.; Jy, W.; Jimenez, J.J.; Sheremata, W.A.; Mauro, L.M.; Mao, W.W.; Horstman, L.L.; Ahn, Y.S. Elevated Plasma Endothelial Microparticles in Multiple Sclerosis. Neurology 2001, 56, 1319–1324. [Google Scholar] [CrossRef]
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for Secretion of Cu,Zn Superoxide Dismutase via Exosomes from a Cell Model of Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2007, 428, 43–46. [Google Scholar] [CrossRef]
- Basso, M.; Pozzi, S.; Tortarolo, M.; Fiordaliso, F.; Bisighini, C.; Pasetto, L.; Spaltro, G.; Lidonnici, D.; Gensano, F.; Battaglia, E.; et al. Mutant Copper-Zinc Superoxide Dismutase (SOD1) Induces Protein Secretion Pathway Alterations and Exosome Release in Astrocytes: Implications for Disease Spreading and Motor Neuron Pathology in Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2013, 288, 15699–15711. [Google Scholar] [CrossRef]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells Release Prions in Association with Exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Chistiakov, A.A. α-Synuclein-Carrying Extracellular Vesicles in Parkinson’s Disease: Deadly Transmitters. Acta Neurol. Belg. 2017, 117, 43–51. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Enasko, J.; Hernandez, C.; Wang, G.; Kong, J.; Helwa, I.; Liu, Y.; Terry, A.V.; Bieberich, E. Neutral Sphingomyelinase-2 Deficiency Ameliorates Alzheimer’s Disease Pathology and Improves Cognition in the 5XFAD Mouse. J. Neurosci. 2016, 36, 8653–8667. [Google Scholar] [CrossRef] [PubMed]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of Conversion from Mild Cognitive Impairment to Dementia with Neuronally Derived Blood Exosome Protein Profile. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Pleet, M.L.; Mathiesen, A.; DeMarino, C.; Akpamagbo, Y.A.; Barclay, R.A.; Schwab, A.; Iordanskiy, S.; Sampey, G.C.; Lepene, B.; Nekhai, S.; et al. Ebola VP40 in Exosomes Can Cause Immune Cell Dysfunction. Front. Microbiol. 2016, 7, 1765. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.-S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 Controls Autophagosome Maturation Essential for Ubiquitin-Selective Quality-Control Autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Mehla, R.; Vijayakumar, T.S.; Handy, I. Endocytosis-Mediated HIV-1 Entry and Its Significance in the Elusive Behavior of the Virus in Astrocytes. Virology 2014, 456, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.A.; Chojnacki, J.; Jones, D.M.; Johnson, E.; Do, T.; Eggeling, C.; Padilla-Parra, S.; Sattentau, Q.J. Astrocytes Resist HIV-1 Fusion but Engulf Infected Macrophage Material. Cell Rep. 2017, 18, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Vijaykumar, T.S.; Nath, A.; Chauhan, A. Chloroquine Mediated Molecular Tuning of Astrocytes for Enhanced Permissiveness to HIV Infection. Virology 2008, 381, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bhakar, A.L.; Tannis, L.-L.; Zeindler, C.; Russo, M.P.; Jobin, C.; Park, D.S.; MacPherson, S.; Barker, P.A. Constitutive Nuclear Factor-Kappa B Activity Is Required for Central Neuron Survival. J. Neurosci. 2002, 22, 8466–8475. [Google Scholar] [CrossRef] [PubMed]
- Koedel, U.; Bayerlein, I.; Paul, R.; Sporer, B.; Pfister, H.W. Pharmacologic Interference with NF-kB Activation Attenuates Central Nervous System Complications in Experimental Pneumococcal Meningitis. J. Infect. Dis. 2000, 182, 1437–1445. [Google Scholar] [CrossRef]
- Kisseleva, T.; Song, L.; Vorontchikhina, M.; Feirt, N.; Kitajewski, J.; Schindler, C. NF-κB Regulation of Endothelial Cell Function during LPS-Induced Toxemia and Cancer. J. Clin. Investig. 2006, 116, 2955–2963. [Google Scholar] [CrossRef]
- Teng, Z.; Guo, Z.; Zhong, J.; Cheng, C.; Huang, Z.; Wu, Y.; Tang, S.; Luo, C.; Peng, X.; Wu, H.; et al. ApoE Influences the Blood-Brain Barrier Through the NF-κB/MMP-9 Pathway After Traumatic Brain Injury. Sci. Rep. 2017, 7, 6649. [Google Scholar] [CrossRef] [PubMed]
- Coelho-Santos, V.; Leitão, R.A.; Cardoso, F.L.; Palmela, I.; Rito, M.; Barbosa, M.; Brito, M.A.; Fontes-Ribeiro, C.A.; Silva, A.P. The TNF-α/NF-κB Signaling Pathway Has a Key Role in Methamphetamine–Induced Blood–Brain Barrier Dysfunction. J. Cereb. Blood Flow Metab. 2015, 35, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, E.; Pacifico, F.; Lavorgna, A.; De Palma, R.; D’Aiuto, E.; Palumbo, G.; Formisano, S.; Leonardi, A. NF-κB-Dependent Cytokine Secretion Controls Fas Expression on Chemotherapy-Induced Premature Senescent Tumor Cells. Oncogene 2011, 30, 2707–2717. [Google Scholar] [CrossRef] [PubMed]
- Rovillain, E.; Mansfield, L.; Caetano, C.; Alvarez-Fernandez, M.; Caballero, O.L.; Medema, R.H.; Hummerich, H.; Jat, P.S. Activation of Nuclear Factor-Kappa B Signalling Promotes Cellular Senescence. Oncogene 2011, 30, 2356–2366. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-κB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Tang, T.M.; Lue, L.-F. Increased Expression of Toll-like Receptor 3, an Anti-Viral Signaling Molecule, and Related Genes in Alzheimer’s Disease Brains. Exp. Neurol. 2018, 309, 91–106. [Google Scholar] [CrossRef]
- McKimmie, C.S.; Fazakerley, J.K. In Response to Pathogens, Glial Cells Dynamically and Differentially Regulate Toll-like Receptor Gene Expression. J. Neuroimmunol. 2005, 169, 116–125. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Geometric Shape Complementarity Score | Interface Area | ACE (kcal/mol) |
|---|---|---|---|
| 1 | 18,466 | 4013.50 | −1492.78 |
| 2 | 17,732 | 3228.50 | −1197.85 |
| 3 | 17,870 | 3453.60 | −1034.96 |
| 4 | 19,060 | 3353.60 | −1009.48 |
| 5 | 18,980 | 3371.30 | −967.94 |
| 6 | 18,072 | 3202.80 | −638.25 |
| 7 | 20,688 | 3171.10 | −624.67 |
| 8 | 18,246 | 3080.00 | −531.61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeMarino, C.; Cowen, M.; Williams, A.; Khatkar, P.; Abulwerdi, F.A.; Henderson, L.; Denniss, J.; Pleet, M.L.; Luttrell, D.R.; Vaisman, I.; et al. Autophagy Deregulation in HIV-1-Infected Cells Increases Extracellular Vesicle Release and Contributes to TLR3 Activation. Viruses 2024, 16, 643. https://doi.org/10.3390/v16040643
DeMarino C, Cowen M, Williams A, Khatkar P, Abulwerdi FA, Henderson L, Denniss J, Pleet ML, Luttrell DR, Vaisman I, et al. Autophagy Deregulation in HIV-1-Infected Cells Increases Extracellular Vesicle Release and Contributes to TLR3 Activation. Viruses. 2024; 16(4):643. https://doi.org/10.3390/v16040643
Chicago/Turabian StyleDeMarino, Catherine, Maria Cowen, Anastasia Williams, Pooja Khatkar, Fardokht A. Abulwerdi, Lisa Henderson, Julia Denniss, Michelle L. Pleet, Delores R. Luttrell, Iosif Vaisman, and et al. 2024. "Autophagy Deregulation in HIV-1-Infected Cells Increases Extracellular Vesicle Release and Contributes to TLR3 Activation" Viruses 16, no. 4: 643. https://doi.org/10.3390/v16040643