
Transcriptomic Investigation of the Virus Spectrum Carried by Midges in Border Areas of Yunnan Province

 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Virus Nucleic Acid Extraction and Gene Library Construction
2.3. Virus Sequencing and Analysis
3. Results
3.1. Sample Collection and Identification
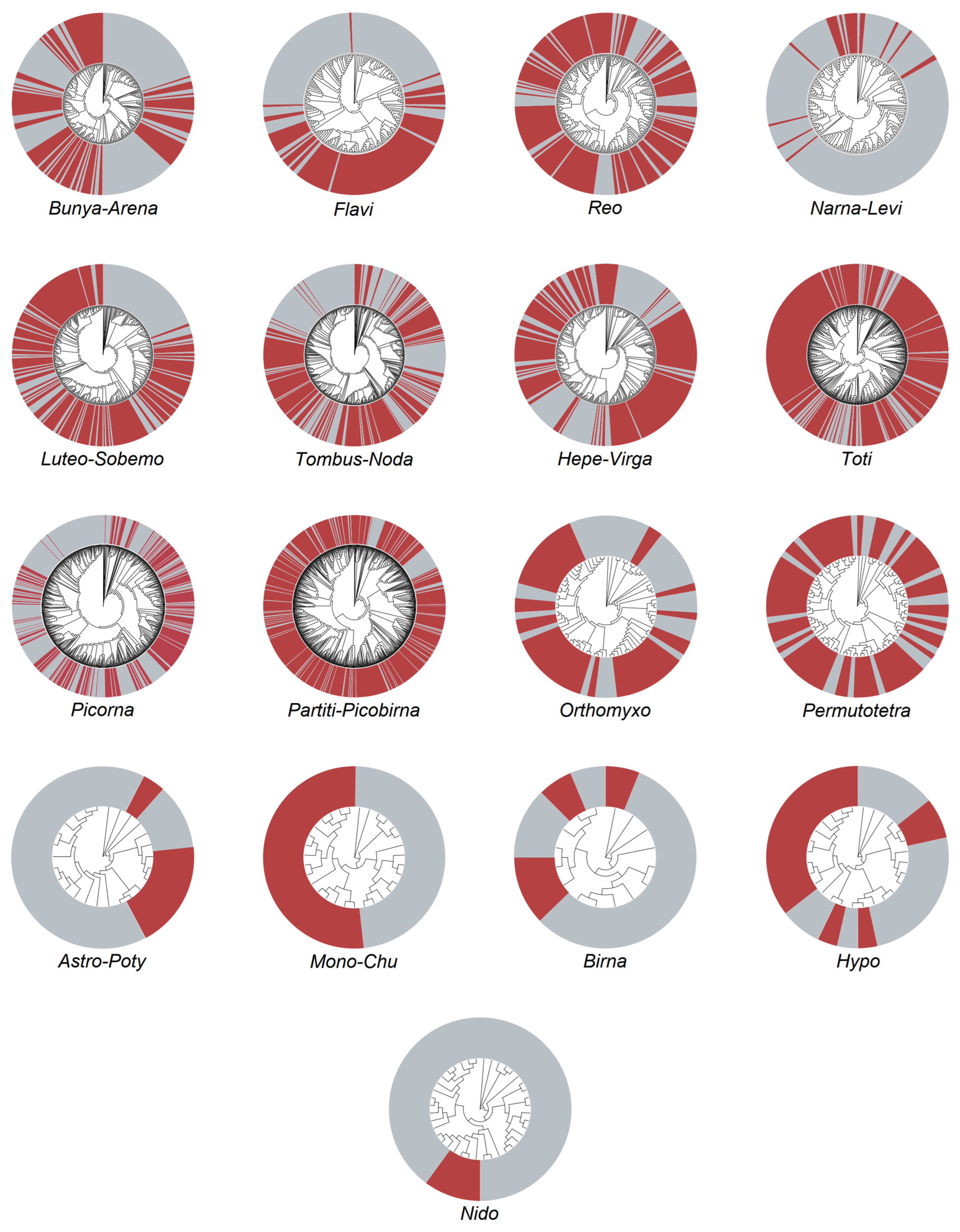
3.2. Viruses Carried by Midges
3.3. Phylogenetic Analysis of Vector-Borne Viruses
4. Discussion
4.1. Diversity of Midge Species
4.2. Viral Diversity
4.3. Diversity of Arboviruses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wagner, R.; Barták, M.; Borkent, A.; Courtney, G.; Goddeeris, B.; Haenni, J.-P.; Knutson, L.; Pont, A.; Rotheray, G.E.; Rozkošný, R.; et al. Global Diversity of Dipteran Families (Insecta diptera) in Freshwater (Excluding Simulidae, Culicidae, Chironomidae, Tipulidae and Tabanidae). Hydrobiologia 2008, 595, 489–519. [Google Scholar] [CrossRef]
- Spinelli, G.R.; Borkent, A.; Ronderos, M.M. Two New Peculiar Species of Neotropical Brachypogon Kieffer (Diptera: Ceratopogonidae). Zootaxa 2013, 3702, 90. [Google Scholar] [CrossRef]
- Foxi, C.; Satta, G.; Puggioni, G.; Ligios, C. Biting Midges (Ceratopogonidae, Culicoides). In Encyclopedia of Infection and Immunity; Elsevier: Amsterdam, The Netherlands, 2022; pp. 852–873. ISBN 978-0-323-90303-5. [Google Scholar]
- Borkent, A.; Wirth, W.W. World Species of Biting Midges (Diptera: Ceratopogonidae); American Museum of Natural History: New York, NY, USA, 1997; Volume 233. [Google Scholar]
- Yu, Y.; Liu, J. World Species of Bloodsucking Midges:(Diptera: Ceratopogonidae); Military Medical Science Press: Beijing, China, 2006; ISBN 7-80121-824-8. [Google Scholar]
- Elbers, A.R.W.; Koenraadt, C.; Meiswinkel, R. Mosquitoes and Culicoides Biting Midges: Vector Range and the Influence of Climate Change: -EN--FR- Les Moustiques et Les Moucherons Piqueurs Culicoides: Diversité Des Vecteurs et Influence Du Changement Climatique -ES- Mosquitos y Jejenes Culicoides: Distribución de Los Vectores e Influencia Del Cambio Climático. Rev. Sci. Tech. OIE 2015, 34, 123–137. [Google Scholar] [CrossRef]
- Blasdell, K.R.; Voysey, R.; Bulach, D.M.; Trinidad, L.; Tesh, R.B.; Boyle, D.B.; Walker, P.J. Malakal Virus from Africa and Kimberley Virus from Australia Are Geographic Variants of a Widely Distributed Ephemerovirus. Virology 2012, 433, 236–244. [Google Scholar] [CrossRef]
- Cybinski, D.H.; St. George, T.D.; Standfast, H.A.; McGregor, A. Isolation of Tibrogargan Virus, a New Australian Rhabdovirus, from Culicoides Brevitarsis. Vet. Microbiol. 1980, 5, 301–308. [Google Scholar] [CrossRef]
- Lei, W.; Guo, X.; Fu, S.; Feng, Y.; Nie, K.; Song, J.; Li, Y.; Ma, X.; Liang, G.; Zhou, H. Isolation of Tibet Orbivirus, TIBOV, from Culicoides Collected in Yunnan, China. PLoS ONE 2015, 10, e0136257. [Google Scholar] [CrossRef]
- Doherty, R.L.D.; Carley, J.G.C.; Standfast, H.A.S.; Dyce, A.L.D.; Snowdon, W.A.S. Virus Strains Isolated from Arthropods during an Epizootic of Bovine Ephemeral Fever in Queensland. Aust. Vet. J. 1972, 48, 81–86. [Google Scholar] [CrossRef]
- Gubala, A.; Davis, S.; Weir, R.; Melville, L.; Cowled, C.; Walker, P.; Boyle, D. Ngaingan Virus, a Macropod-Associated Rhabdovirus, Contains a Second Glycoprotein Gene and Seven Novel Open Reading Frames. Virology 2010, 399, 98–108. [Google Scholar] [CrossRef]
- Karabatsos, N. Supplement to International Catalogue of Arboviruses Including Certain Other Viruses of Vertebrates. Am. J. Trop. Med. Hyg. 1978, 27, 372. [Google Scholar] [CrossRef]
- Mellor, P.S.; Hamblin, C. African Horse Sickness. Vet. Res. 2004, 35, 445–466. [Google Scholar] [CrossRef]
- Pilo, P. Phylogenetic Lineages of Francisella Tularensis in Animals. Front. Cell. Infect. Microbiol. 2018, 8, 258. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Y.; Wang, J.-S. Culicoides Arakawae (Diptera: Ceratopogonidae) Efficiently Blood-Fed and Infected with Leucocytozoon Caulleryi through a Natural Membrane. Vet. Parasitol. 2001, 99, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Hardikar, A.A.; Nath, B.B. Chromosomal Polymorphism Is Associated with Nematode Parasitism in a Natural Population of a Tropical Midge. Chromosoma 2001, 110, 58–64. [Google Scholar] [CrossRef]
- Weaver, S.C.; Reisen, W.K. Present and Future Arboviral Threats. Antivir. Res. 2010, 85, 328–345. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Takasaki, T.; Fu, S.H.; Sun, X.H.; Zhang, H.L.; Wang, Z.X.; Hao, Z.Y.; Zhang, J.K.; Tang, Q.; Kotaki, A.; et al. Molecular Epidemiological Analysis of Japanese Encephalitis Virus in China. J. Gen. Virol. 2007, 88, 885–894. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; He, Y.; Zhou, Y.; Xin, A.; Liao, D.; Meng, J. Isolation of Tibet Orbivirus from Culicoides and Associated Infections in Livestock in Yunnan, China. Virol. J. 2017, 14, 105. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.L.; Bellis, G.; Li, L.; Li, H.C.; Miao, H.S.; Kou, M.L.; Liao, D.F.; Wang, Z.; Gao, L.; Li, J.Z. Potential Vectors of Bluetongue Virus in High Altitude Areas of Yunnan Province, China. Parasites Vectors 2019, 12, 464. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, X.; Li, Y.; Hu, N.; Sun, J.; Wu, M.; Zhou, H.; Hu, Y. Evolutionary Analysis of a Newly Isolated Banna Virus Strain from Yunnan, China. Arch. Virol. 2022, 167, 1221–1223. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the Invertebrate RNA Virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Schuh, A.J.; Ward, M.J.; Leigh Brown, A.J.; Barrett, A.D.T. Dynamics of the Emergence and Establishment of a Newly Dominant Genotype of Japanese Encephalitis Virus throughout Asia. J. Virol. 2014, 88, 4522–4532. [Google Scholar] [CrossRef]
- Duong, V.; Choeung, R.; Gorman, C.; Laurent, D.; Crabol, Y.; Mey, C.; Peng, B.; Di Francesco, J.; Hul, V.; Sothy, H.; et al. Isolation and Full-Genome Sequences of Japanese Encephalitis Virus Genotype I Strains from Cambodian Human Patients, Mosquitoes and Pigs. J. Gen. Virol. 2017, 98, 2287–2296. [Google Scholar] [CrossRef]
- Borkent, A.; Dominiak, P. Catalog of the Biting Midges of the World (Diptera: Ceratopogonidae). Zootaxa 2020, 4787, 1–377. [Google Scholar] [CrossRef]
- Tabachnick, W.J. Genetic Differentiation Among Populations of Culicoides Variipennis (Diptera: Ceratopogonidae), the North American Vector of Bluetongue Virus. Ann. Entomol. Soc. Am. 1992, 85, 140–147. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Ratnasingham, S.; De Waard, J.R. Barcoding Animal Life: Cytochrome c Oxidase Subunit 1 Divergences among Closely Related Species. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, S96–S99. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Penton, E.H.; Burns, J.M.; Janzen, D.H.; Hallwachs, W. Ten Species in One: DNA Barcoding Reveals Cryptic Species in the Neotropical Skipper Butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA 2004, 101, 14812–14817. [Google Scholar] [CrossRef] [PubMed]
- Pagès, N.; Monteys, V.S. i Differentiation of Culicoides Obsoletus and Culicoides Scoticus (Diptera: Ceratopogonidae) Based on Mitochondrial Cytochrome Oxidase Subunit I. J. Med. Entomol. 2005, 42, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.P.; Guo, X.F.; Li, Y.Y.; Zhang, J.; Wang, J.; Li, C.M.; Yang, Z.M.; Chen, H.Y.; Zhou, H.N.; Liang, G.D. Survey of Hematophagous Midges in China-Laos Border. Chin. J. Vector Biol. Control 2016, 27, 463–466. [Google Scholar] [CrossRef]
- Di, D.; Li, C.; Li, Z.; Wang, X.; Xia, Q.; Sharma, M.; Li, B.; Liu, K.; Shao, D.; Qiu, Y.; et al. Detection of Arboviruses in Culicoides (Diptera: Ceratopogonidae) Collected from Animal Farms in the Border Areas of Yunnan Province, China. J. Integr. Agric. 2021, 20, 2491–2501. [Google Scholar] [CrossRef]
- Wang, F.; Yu, Y.; Ouyang, M. Four New Record Species of the Genus Culicoides (Diptera, Ceratopogonidae) from China. Acta Zootaxon. Sin./Dongwu Fenlei Xuebao 2013, 38, 922–924. [Google Scholar]
- Webster, C.L.; Waldron, F.M.; Robertson, S.; Crowson, D.; Ferrari, G.; Quintana, J.F.; Brouqui, J.-M.; Bayne, E.H.; Longdon, B.; Buck, A.H.; et al. The Discovery, Distribution, and Evolution of Viruses Associated with Drosophila Melanogaster. PLoS Biol. 2015, 13, e1002210. [Google Scholar] [CrossRef]
- Xu, P.; Yang, L.; Yang, X.; Li, T.; Graham, R.I.; Wu, K.; Wilson, K. Novel Partiti-like Viruses Are Conditional Mutualistic Symbionts in Their Normal Lepidopteran Host, African Armyworm, but Parasitic in a Novel Host, Fall Armyworm. PLoS Pathog. 2020, 16, e1008467. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Chen, Y.; Wei, X.; Cui, J. Viromes in Marine Ecosystems Reveal Remarkable Invertebrate RNA Virus Diversity. Sci. China Life Sci. 2022, 65, 426–437. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus Taxonomy: The Database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef]
- Tang, S.; Shen, S.; Shi, J.; Fang, Y.; Wang, H.; Hu, Z.; Deng, F. A Review and Novel Classification of Bunyavirales. Biodivers. Sci. 2018, 26, 1004. [Google Scholar] [CrossRef]
- Gao, X.; Nasci, R.; Liang, G. The Neglected Arboviral Infections in Mainland China. PLoS Neglected Trop. Dis. 2010, 4, e624. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Zhou, H. The Spatiotemporal Distribution of Japanese Encephalitis Cases in Yunnan Province, China, from 2007 to 2017. PLoS ONE 2020, 15, e0231661. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.-L.; Liu, H.; Wang, H.-Y.; Fu, S.-H.; Liu, H.-Z.; Zhang, H.-L.; Li, M.-H.; Gao, X.-Y.; Wang, J.-L.; Sun, X.-H.; et al. Emergence of Genotype I of Japanese Encephalitis Virus as the Dominant Genotype in Asia. J. Virol. 2011, 85, 9847–9853. [Google Scholar] [CrossRef]
- Xie, J.; Miao, H.; Kou, M.; Li, H. Antibody Detection of Akabane Disease Virus in Border Areas of Yunnan Province from 2017 to 2018. China Acad. J. Electron. Publ. House 2019, 36, 23–28. [Google Scholar] [CrossRef]
- Feng, Y.; Fu, S.; Zhang, H.; Li, M.; Zhou, T.; Wang, J.; Zhang, Y.; Wang, H.; Tang, Q.; Liang, G. Distribution of Mosquitoes and Mosquito-Borne Viruses along the China-Myanmar Border in Yunnan Province. Jpn. J. Infect. Dis. 2012, 65, 215–221. [Google Scholar] [CrossRef]
- Xiao, P.; Han, J.; Zhang, Y.; Li, C.; Guo, X.; Wen, S.; Tian, M.; Li, Y.; Wang, M.; Liu, H.; et al. Metagenomic Analysis of Flaviviridae in Mosquito Viromes Isolated from Yunnan Province in China Reveals Genes from Dengue and Zika Viruses. Front. Cell. Infect. Microbiol. 2018, 8, 359. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, W.; Xue, J.-B.; Zhang, Y. Monitoring Mosquito-Borne Arbovirus in Various Insect Regions in China in 2018. Front. Cell. Infect. Microbiol. 2021, 11, 640993. [Google Scholar] [CrossRef]
- Xu, Z.; Feng, Y.; Chen, X.; Shi, M.; Fu, S.; Yang, W.; Liu, W.J.; Gao, G.F.; Liang, G. Virome of Bat-Infesting Arthropods: Highly Divergent Viruses in Different Vectors. J. Virol. 2022, 96, e01464-21. [Google Scholar] [CrossRef]
- Tian, F.; He, J.; Shang, S.; Chen, Z.; Tang, Y.; Lu, M.; Huang, C.; Guo, X.; Tong, Y. Survey of Mosquito Species and Mosquito-Borne Viruses in Residential Areas along the Sino–Vietnam Border in Yunnan Province in China. Front. Microbiol. 2023, 14, 1105786. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Wu, W.; Cai, S.; Wang, J.; Kuang, G.; Yang, W.; Wang, J.; Han, X.; Pan, H.; Shi, M.; et al. Transcriptomic Investigation of the Virus Spectrum Carried by Midges in Border Areas of Yunnan Province. Viruses 2024, 16, 674. https://doi.org/10.3390/v16050674
Yang L, Wu W, Cai S, Wang J, Kuang G, Yang W, Wang J, Han X, Pan H, Shi M, et al. Transcriptomic Investigation of the Virus Spectrum Carried by Midges in Border Areas of Yunnan Province. Viruses. 2024; 16(5):674. https://doi.org/10.3390/v16050674
Chicago/Turabian StyleYang, Lifen, Weichen Wu, Sa Cai, Jing Wang, Guopeng Kuang, Weihong Yang, Juan Wang, Xi Han, Hong Pan, Mang Shi, and et al. 2024. "Transcriptomic Investigation of the Virus Spectrum Carried by Midges in Border Areas of Yunnan Province" Viruses 16, no. 5: 674. https://doi.org/10.3390/v16050674