
Innate Immune Evasion of PRRSV nsp11 through Degradation of the HDAC2 by Its Endoribonuclease Activity
 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viruses
2.2. Constructs and Antibodies
2.3. Virus Infection and Cell Transfection
2.4. RNA Interference Assay
2.5. Western Blotting Assay
2.6. Quantitative Reverse Transcription-PCR (RT-qPCR)
2.7. TCID50 Assay
2.8. CCK-8 Assay
2.9. Statistical Analysis
3. Results
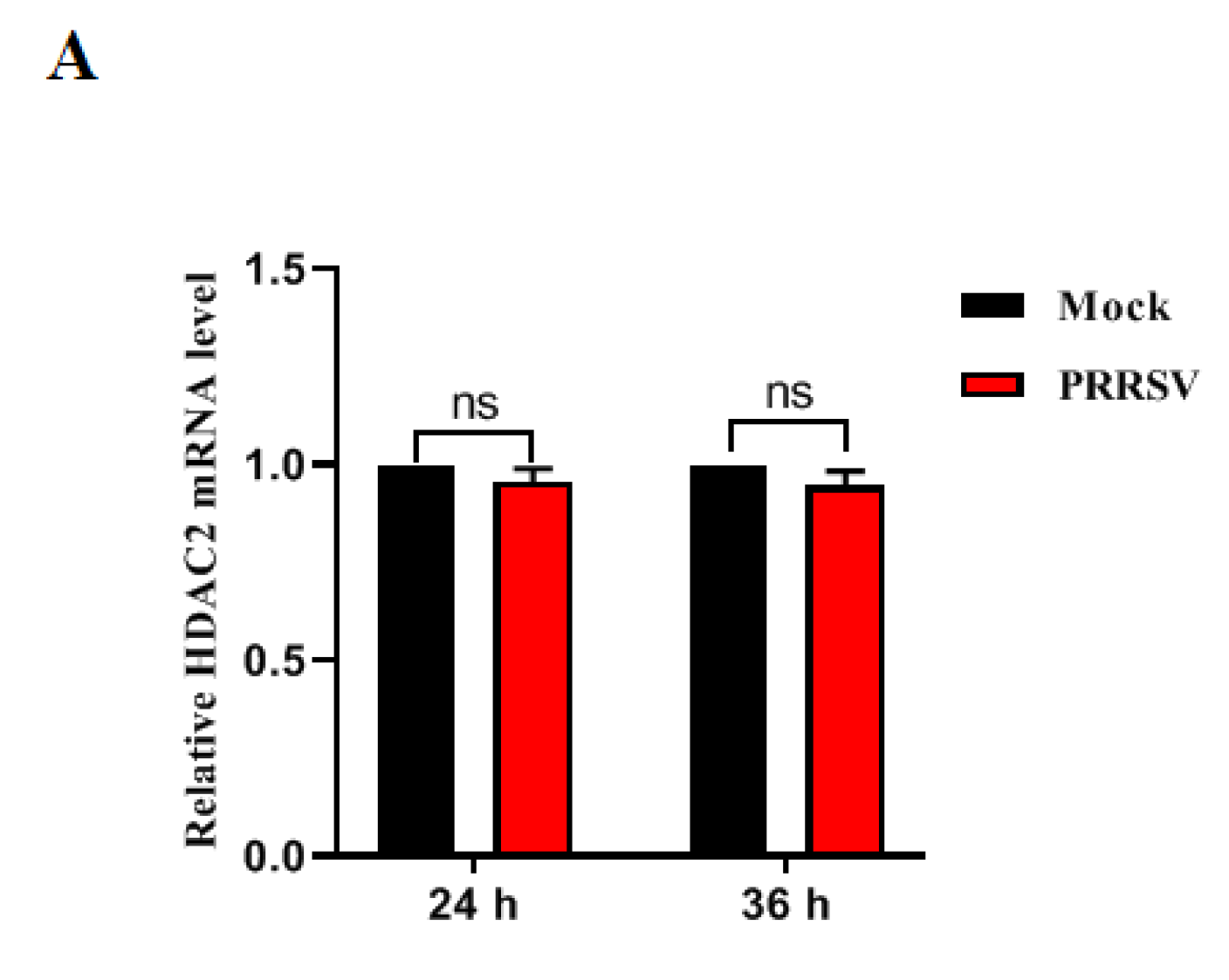
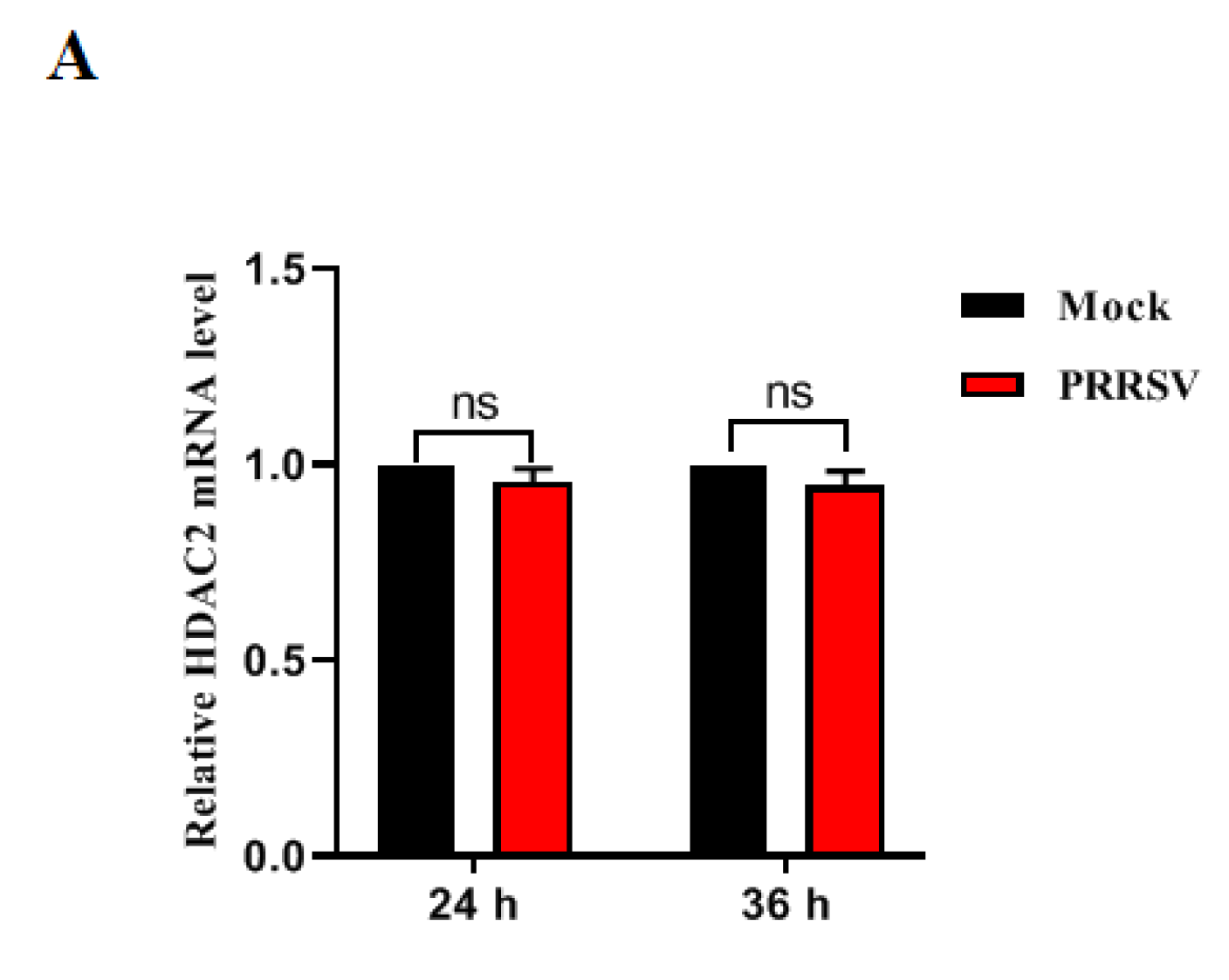
3.1. PRRSV Infection Downregulates HDAC2 Expression
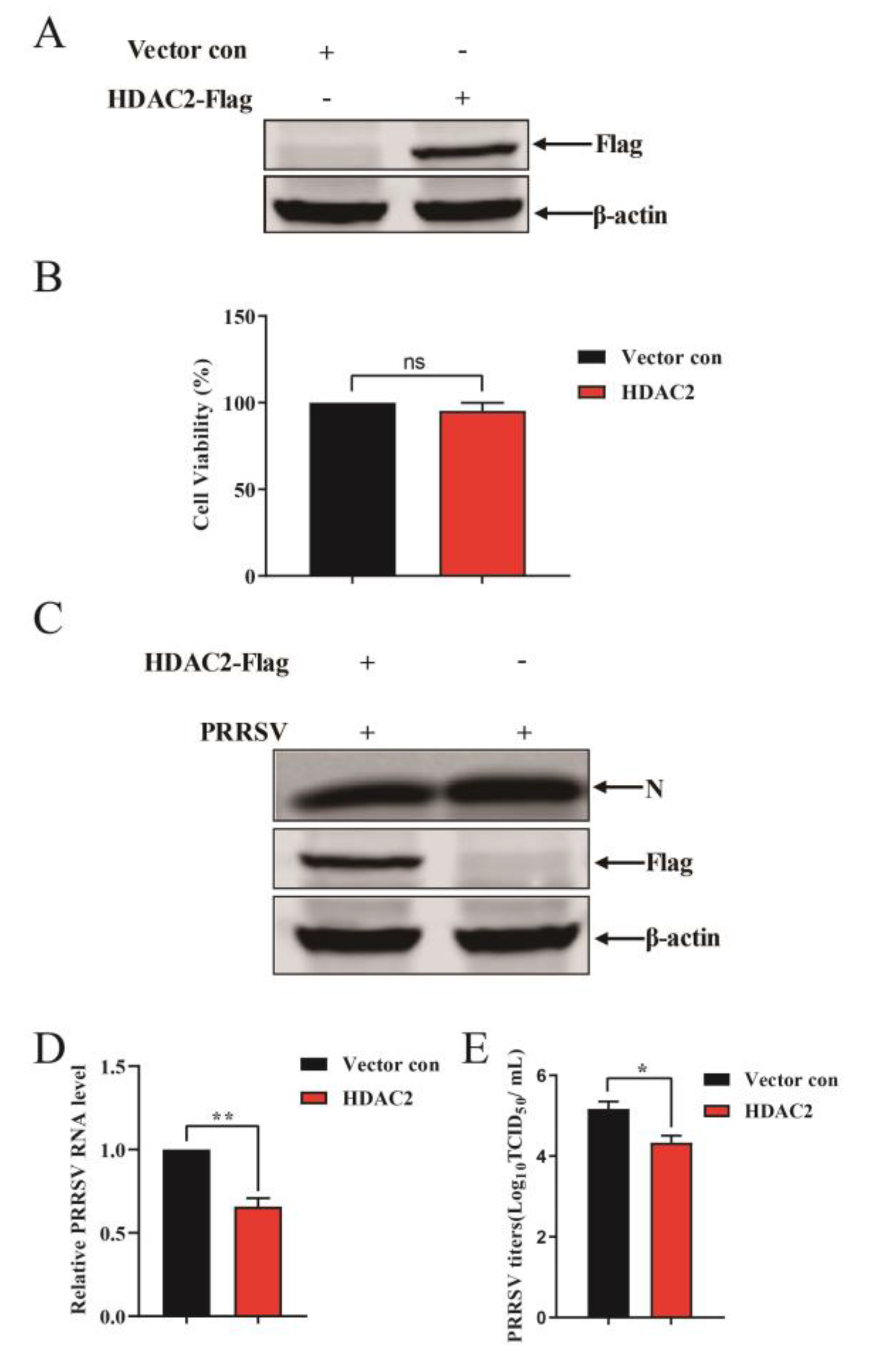
3.2. HDAC2 Negatively Regulates PRRSV Replication
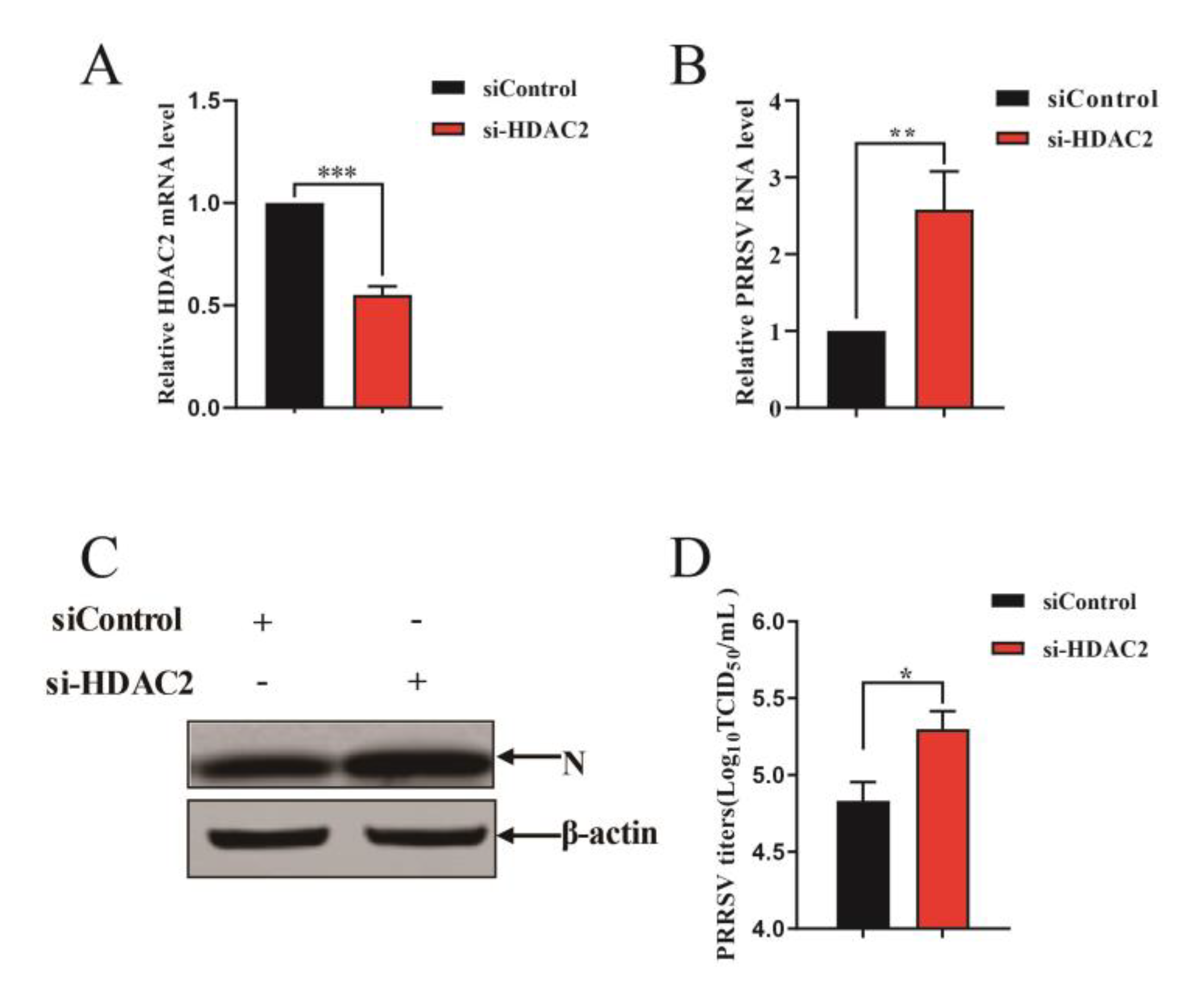
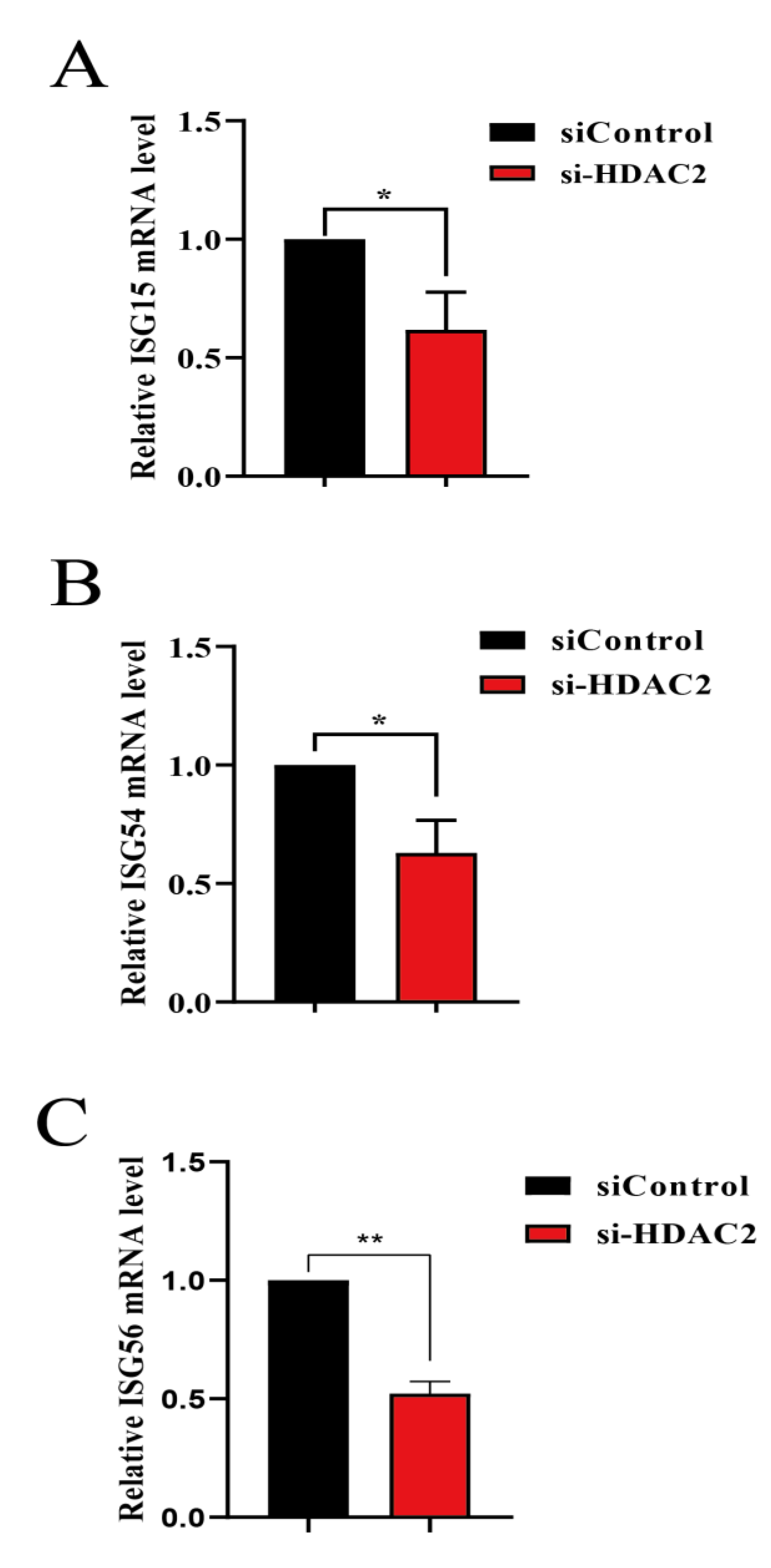
3.3. Knockdown of Endogenous HDAC2 Promotes PRRSV Infection
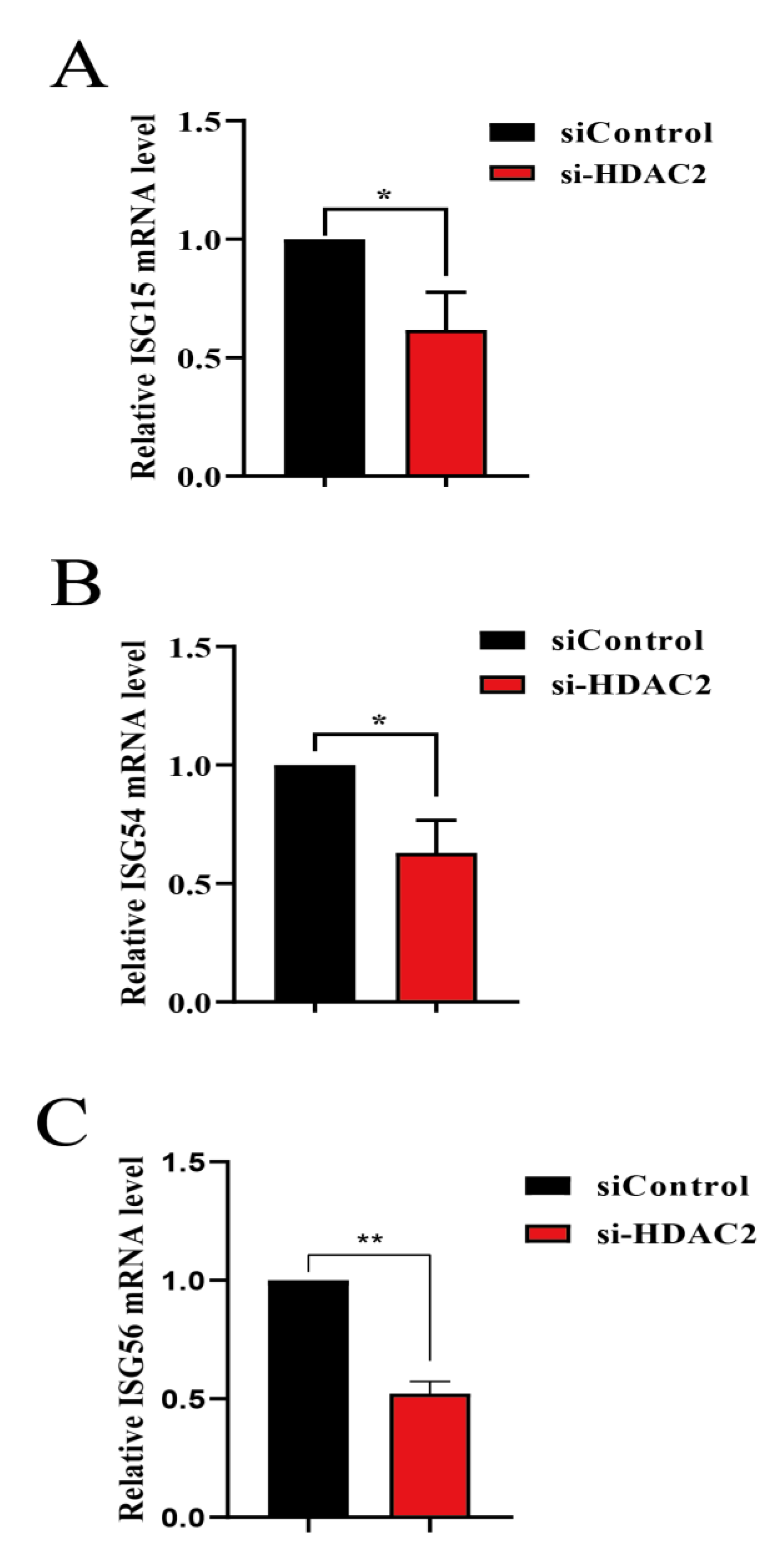
3.4. HDAC2 Positively Regulates IFN Antiviral Responses
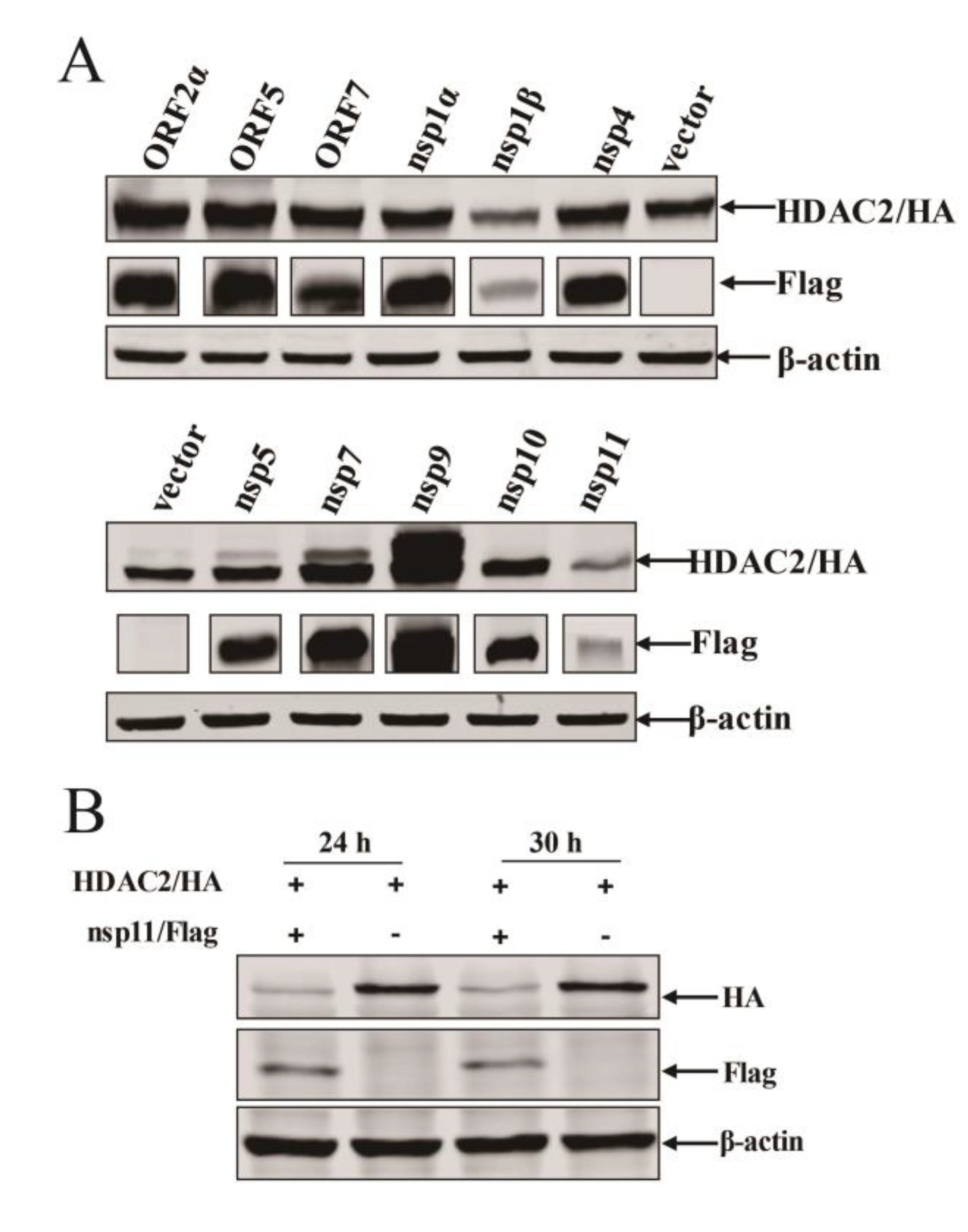
3.5. PRRSV nsp11 Inhibits HDAC2 Expression
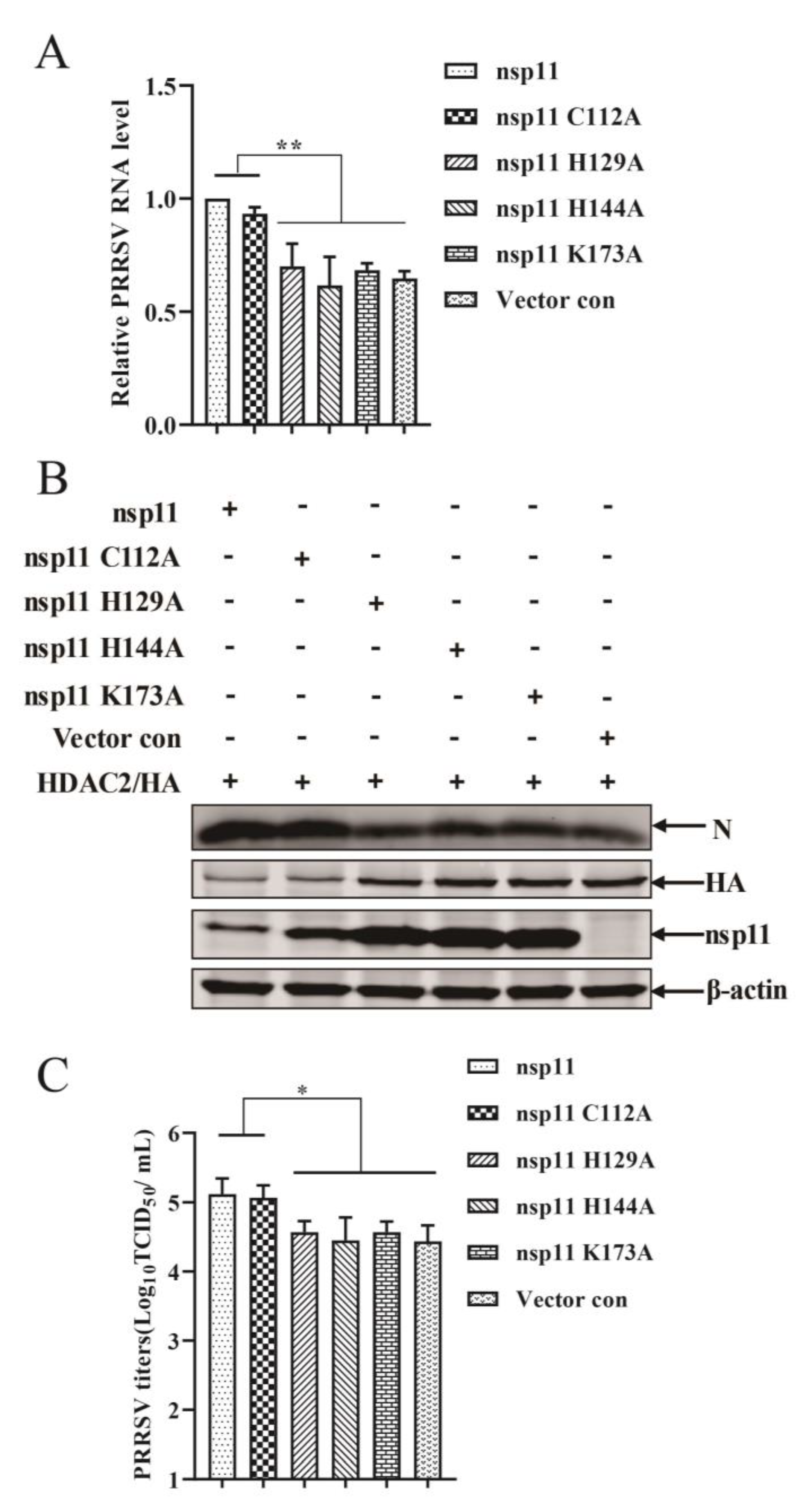
3.6. nsp11 Mediated HDAC2 Reduction in an Endonuclease Activity Dependent Manner
3.7. PRRSV nsp11 Antagonizes the Antiviral Response of HDAC2 in an Endonuclease Activity Dependent Manner
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nelsen, C.J.; Murtaugh, M.P.; Faaberg, K.S. Porcine reproductive and respiratory syndrome virus comparison: Divergent evolution on two continents. J. Virol. 1999, 73, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Ruedas-Torres, I.; Rodriguez-Gomez, I.M.; Sanchez-Carvajal, J.M.; Larenas-Munoz, F.; Pallares, F.J.; Carrasco, L.; Gomez-Laguna, J. The jigsaw of PRRSV virulence. Vet. Microbiol. 2021, 260, 109168. [Google Scholar] [CrossRef] [PubMed]
- Yim-Im, W.; Anderson, T.K.; Paploski, I.A.D.; VanderWaal, K.; Gauger, P.; Krueger, K.; Shi, M.; Main, R.; Zhang, J. Refining PRRSV-2 genetic classification based on global ORF5 sequences and investigation of their geographic distributions and temporal changes. Microbiol. Spectr. 2023, 11, e0291623. [Google Scholar] [CrossRef]
- Snijder, E.J.; Kikkert, M.; Fang, Y. Arterivirus molecular biology and pathogenesis. J. Gen. Virol. 2013, 94(Pt. 10), 2141–2163. [Google Scholar] [CrossRef] [PubMed]
- Dokland, T. The structural biology of PRRSV. Virus Res. 2010, 154, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Lugo Mesa, V.; Quinonez Munoz, A.; Sobhy, N.M.; Corzo, C.A.; Goyal, S.M. Survival of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) in the Environment. Vet. Sci. 2024, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Papatsiros, V.; Stylianaki, I.; Papakonstantinou, G.; Tsekouras, N.; Bitchava, D.; Christodoulopoulos, G.; Papaioannou, N. Histopathological Lesions Accompanied with First-Time Isolation of a PRRSV-2 Strain in Greece. Viral Immunol. 2020, 33, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Wellenberg, G.J. Review: Diagnostic methods for the detection of porcine reproductive and respiratory syndrome virus (PRRSV) infections. Tijdschr. Voor Diergeneeskd. 2006, 131, 566–572. [Google Scholar]
- Yuzhakov, A.G.; Raev, S.A.; Shchetinin, A.M.; Gushchin, V.A.; Alekseev, K.P.; Stafford, V.V.; Komina, A.K.; Zaberezhny, A.D.; Gulyukin, A.M.; Aliper, T.I. Full-genome analysis and pathogenicity of a genetically distinct Russian PRRSV-1 Tyu16 strain. Vet. Microbiol. 2020, 247, 108784. [Google Scholar] [CrossRef]
- Sinkora, M.; Toman, M.; Stepanova, K.; Stepanova, H.; Leva, L.; Sinkorova, J.; Moutelikova, R.; Salat, J.; Srutkova, D.; Schwarzer, M.; et al. The mechanism of immune dysregulation caused by porcine reproductive and respiratory syndrome virus (PRRSV). Microbes Infect. 2023, 25, 105146. [Google Scholar] [CrossRef]
- Fiers, J.; Tignon, M.; Maes, D.; Cay, A.B. Follow-Up of PRRSv-Vaccinated Piglets Born from PRRSv-Vaccinated, ELISA-Seropositive and ELISA-Seronegative Sows. Viruses 2023, 15, 479. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Qiao, S.; Li, R.; Wang, J.; Li, X.; Zhang, G. Evasion strategies of porcine reproductive and respiratory syndrome virus. Front Microbiol. 2023, 14, 1140449. [Google Scholar] [CrossRef]
- Kim, O.; Sun, Y.; Lai, F.W.; Song, C.; Yoo, D. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells. Virology 2010, 402, 315–326. [Google Scholar] [CrossRef]
- Han, M.; Du, Y.; Song, C.; Yoo, D. Degradation of CREB-binding protein and modulation of type I interferon induction by the zinc finger motif of the porcine reproductive and respiratory syndrome virus nsp1alpha subunit. Virus Res. 2013, 172, 54–65. [Google Scholar] [CrossRef]
- Beura, L.K.; Sarkar, S.N.; Kwon, B.; Subramaniam, S.; Jones, C.; Pattnaik, A.K.; Osorio, F.A. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J. Virol. 2010, 84, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Frias-Staheli, N.; Giannakopoulos, N.V.; Kikkert, M.; Taylor, S.L.; Bridgen, A.; Paragas, J.; Richt, J.A.; Rowland, R.R.; Schmaljohn, C.S.; Lenschow, D.J.; et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2007, 2, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, D.; Sun, Z.; Gao, L.; Zhu, X.; Guo, J.; Xu, S.; Fang, L.; Li, K.; Xiao, S. Arterivirus nsp4 Antagonizes Interferon Beta Production by Proteolytically Cleaving NEMO at Multiple Sites. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; He, J.; Wang, R.; Zhang, X.; Lin, S.; Ma, Z.; Zhang, Y. Nonstructural Protein 11 of Porcine Reproductive and Respiratory Syndrome Virus Induces STAT2 Degradation to Inhibit Interferon Signaling. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Zhao, K.; Li, L.W.; Jiang, Y.F.; Gao, F.; Zhang, Y.J.; Zhao, W.Y.; Li, G.X.; Yu, L.X.; Zhou, Y.J.; Tong, G.Z. Nucleocapsid protein of porcine reproductive and respiratory syndrome virus antagonizes the antiviral activity of TRIM25 by interfering with TRIM25-mediated RIG-I ubiquitination. Vet. Microbiol. 2019, 233, 140–146. [Google Scholar] [CrossRef]
- Zhao, Y.; Song, Z.; Bai, J.; Liu, X.; Nauwynck, H.; Jiang, P. Porcine reproductive and respiratory syndrome virus Nsp4 cleaves ZAP to antagonize its antiviral activity. Vet. Microbiol. 2020, 250, 108863. [Google Scholar] [CrossRef]
- Tao, R.; Fang, L.; Bai, D.; Ke, W.; Zhou, Y.; Wang, D.; Xiao, S. Porcine Reproductive and Respiratory Syndrome Virus Nonstructural Protein 4 Cleaves Porcine DCP1a To Attenuate Its Antiviral Activity. J. Immunol. 2018, 201, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, C.; Zhou, L.; Zhang, N.; Wang, X.; Ge, X.; Guo, X.; Yang, H. Porcine reproductive and respiratory syndrome virus counteracts the porcine intrinsic virus restriction factors-IFITM1 and Tetherin in MARC-145 cells. Virus Res. 2014, 191, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ke, W.; Fang, L.; Tao, R.; Li, Y.; Jing, H.; Wang, D.; Xiao, S. Porcine Reproductive and Respiratory Syndrome Virus E Protein Degrades Porcine Cholesterol 25-Hydroxylase via the Ubiquitin-Proteasome Pathway. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, Y.; Zhao, W.; Liu, J.; Ullah, R.; Fang, P.; Fang, L.; Xiao, S. Porcine reproductive and respiratory syndrome virus degrades DDX10 via SQSTM1/p62-dependent selective autophagy to antagonize its antiviral activity. Autophagy 2023, 19, 2257–2274. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Naik, N.G.; Lee, S.C.; Veronese, B.H.S.; Ma, Z.; Toth, Z. Interaction of HDAC2 with SARS-CoV-2 NSP5 and IRF3 Is Not Required for NSP5-Mediated Inhibition of Type I Interferon Signaling Pathway. Microbiol. Spectr. 2022, 10, e0232222. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.T.; Hussain, M.; Galvin, H.D.; Husain, M. Histone Deacetylase 2 Is a Component of Influenza A Virus-Induced Host Antiviral Response. Front. Microbiol. 2017, 8, 1315. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Shim, K.; Kim, H.U.; Jung, H.S.; Jeoung, D. HDAC2 as a target for developing anti-cancer drugs. Comput. Struct. Biotechnol. J. 2023, 21, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Liu, Y.G.; Li, L.; Wang, G.; Wang, H.M.; Zhang, H.L.; Zhao, S.F.; Gao, J.C.; An, T.Q.; Tian, Z.J.; et al. Porcine alveolar macrophage CD163 abundance is a pivotal switch for porcine reproductive and respiratory syndrome virus infection. Oncotarget 2018, 9, 12174–12185. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, M.; Zhang, L.; Pan, Y.; Zhang, W.; Ma, W.; Chen, H.; Tang, L.; Xia, C.; Wang, Y. Glycoprotein Non-Metastatic Melanoma Protein B Restricts PRRSV Replication by Inhibiting Autophagosome-Lysosome Fusion. Viruses 2023, 15, 920. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, Y.; Xu, Y.; Zhang, W.; Ma, W.; Ibrahim, Y.M.; Werid, G.M.; Zhang, H.; Xia, C.; Wei, P.; et al. Paraoxonase-1 Facilitates PRRSV Replication by Interacting with Viral Nonstructural Protein-9 and Inhibiting Type I Interferon Pathway. Viruses 2022, 14, 1203. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 365300. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef]
- Talbot-Cooper, C.; Pantelejevs, T.; Shannon, J.P.; Cherry, C.R.; Au, M.T.; Hyvonen, M.; Hickman, H.D.; Smith, G.L. Poxviruses and paramyxoviruses use a conserved mechanism of STAT1 antagonism to inhibit interferon signaling. Cell Host Microbe 2022, 30, 357–372. [Google Scholar] [CrossRef]
- Brockmeier, S.L.; Loving, C.L.; Eberle, K.C.; Hau, S.J.; Buckley, A.; Van Geelen, A.; Montiel, N.A.; Nicholson, T.; Lager, K.M. Interferon alpha inhibits replication of a live-attenuated porcine reproductive and respiratory syndrome virus vaccine preventing development of an adaptive immune response in swine. Vet. Microbiol. 2017, 212, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, L.; Pan, Y.; Gao, J.; Xu, Y.; Li, X.; Tian, Z.; Chen, H.; Wang, Y. Downregulation of miR-218 by porcine reproductive and respiratory syndrome virus facilitates viral replication via inhibition of type I interferon responses. J. Biol. Chem. 2021, 296, 100683. [Google Scholar] [CrossRef]
- Yoo, D.; Song, C.; Sun, Y.; Du, Y.; Kim, O.; Liu, H.C. Modulation of host cell responses and evasion strategies for porcine reproductive and respiratory syndrome virus. Virus Res. 2010, 154, 48–60. [Google Scholar] [CrossRef]
- Luo, R.; Xiao, S.; Jiang, Y.; Jin, H.; Wang, D.; Liu, M.; Chen, H.; Fang, L. Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-beta production by interfering with the RIG-I signaling pathway. Mol. Immunol. 2008, 45, 2839–2846. [Google Scholar] [CrossRef]
- Contreras-Luna, M.J.; Fragoso-Gonzalez, G.; Segura-Velazquez, R.A.; Cervantes-Torres, J.B.; Alonso-Morales, R.; Ramirez-Martinez, L.A.; Ayon-Nunez, D.A.; Bobes, R.J.; Sanchez-Betancourt, J.I. Immunogenic and antigenic analysis of recombinant NSP1 and NSP11 of PRRS virus. Vet. Med. Sci. 2022, 8, 610–618. [Google Scholar] [CrossRef]
- Nedialkova, D.D.; Ulferts, R.; van den Born, E.; Lauber, C.; Gorbalenya, A.E.; Ziebuhr, J.; Snijder, E.J. Biochemical characterization of arterivirus nonstructural protein 11 reveals the nidovirus-wide conservation of a replicative endoribonuclease. J. Virol. 2009, 83, 5671–5682. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, X.; Chang, Y.; Jiang, B.; Deng, R.; Wang, A.; Zhang, G. Nonstructural protein 11 (nsp11) of porcine reproductive and respiratory syndrome virus (PRRSV) promotes PRRSV infection in MARC-145 cells. BMC Vet. Res. 2016, 12, 90. [Google Scholar] [CrossRef]
- Rascon-Castelo, E.; Burgara-Estrella, A.; Mateu, E.; Hernandez, J. Immunological features of the non-structural proteins of porcine reproductive and respiratory syndrome virus. Viruses 2015, 7, 873–886. [Google Scholar] [CrossRef]
- Sun, Y.; Ke, H.; Han, M.; Chen, N.; Fang, W.; Yoo, D. Nonstructural Protein 11 of Porcine Reproductive and Respiratory Syndrome Virus Suppresses Both MAVS and RIG-I Expression as One of the Mechanisms to Antagonize Type I Interferon Production. PLoS ONE 2016, 11, e0168314. [Google Scholar] [CrossRef]
- Shi, X.; Wang, L.; Li, X.; Zhang, G.; Guo, J.; Zhao, D.; Chai, S.; Deng, R. Endoribonuclease activities of porcine reproductive and respiratory syndrome virus nsp11 was essential for nsp11 to inhibit IFN-β induction. Mol. Immunol. 2011, 48, 1568–1572. [Google Scholar] [CrossRef]
- Jiang, D.; He, M.; Sui, C.; Wu, X.; Hu, Y.; Cong, X.; Li, J.; Du, Y.; Qi, J. PRRSV nonstructural protein 11 degrades swine ISG15 by its endoribonuclease activity to antagonize antiviral immune response. Vet. Microbiol. 2023, 280, 109720. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fan, J.; Fang, L.; Luo, R.; Ouyang, H.; Ouyang, C.; Zhang, H.; Chen, H.; Li, K.; Xiao, S. The nonstructural protein 11 of porcine reproductive and respiratory syndrome virus inhibits NF-κB signaling by means of its deubiquitinating activity. Mol. Immunol. 2015, 68 Pt A, 357–366. [Google Scholar] [CrossRef]
- Su, Y.; Shi, P.; Zhang, L.; Lu, D.; Zhao, C.; Li, R.; Zhang, L.; Huang, J. The Superimposed Deubiquitination Effect of OTULIN and Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Nsp11 Promotes Multiplication of PRRSV. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. Porcine reproductive and respiratory syndrome virus nsp1beta and nsp11 antagonize the antiviral activity of cholesterol-25-hydroxylase via lysosomal degradation. Vet. Microbiol. 2018, 223, 134–143. [Google Scholar] [CrossRef]
- Wang, D.; Chen, J.; Yu, C.; Zhu, X.; Xu, S.; Fang, L.; Xiao, S. Porcine Reproductive and Respiratory Syndrome Virus nsp11 Antagonizes Type I Interferon Signaling by Targeting IRF9. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, F.; Li, L.; Zhao, K.; Jiang, S.; Jiang, Y.; Yu, L.; Zhou, Y.; Liu, C.; Tong, G. Porcine Reproductive and Respiratory Syndrome Virus Antagonizes PCSK9’s Antiviral Effect via Nsp11 Endoribonuclease Activity. Viruses 2020, 12, 655. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| HDAC2 | TTTGGTACCATGGCGTACAGTCAGGGAGGCGG | TTTCTCGAGTCAAGGGTTGCTGAGCTGTTCTGA |
| qHDAC2 | CTTGCCATCCTTGAGTTA | TTTAGCGTGACCTTTGAC |
| qPRRSV-ORF7 | AGATCATCGCCCAACAAAAAC | GACACAATTGCCGCTCACTA |
| qISG15 | CCTGTTGATGGTGCAAAGCT | TGCACATAGGCTTGAGGTCA |
| qISG54 | CATTGACCCTCTGAGGCAAG | AGCGTGTCCTATTAGTTCC |
| qISG56 | CATACATTTCCACTATGG | TACTCCAGGGCTTCATTCA |
| qβ-actin | CTTCCTGGGCATGGAGTCC | GGCGCGATGATCTTGATCTTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Chen, J.; Yu, C.; Pan, Y.; Ma, W.; Feng, H.; Xie, J.; Chen, H.; Wang, Y.; Xia, C. Innate Immune Evasion of PRRSV nsp11 through Degradation of the HDAC2 by Its Endoribonuclease Activity. Viruses 2024, 16, 678. https://doi.org/10.3390/v16050678
Zhang H, Chen J, Yu C, Pan Y, Ma W, Feng H, Xie J, Chen H, Wang Y, Xia C. Innate Immune Evasion of PRRSV nsp11 through Degradation of the HDAC2 by Its Endoribonuclease Activity. Viruses. 2024; 16(5):678. https://doi.org/10.3390/v16050678
Chicago/Turabian StyleZhang, He, Jianxing Chen, Changqing Yu, Yu Pan, Wenjie Ma, Hao Feng, Jinxin Xie, Hongyan Chen, Yue Wang, and Changyou Xia. 2024. "Innate Immune Evasion of PRRSV nsp11 through Degradation of the HDAC2 by Its Endoribonuclease Activity" Viruses 16, no. 5: 678. https://doi.org/10.3390/v16050678