Innate Antiviral Immune Responses to Hepatitis B Virus



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
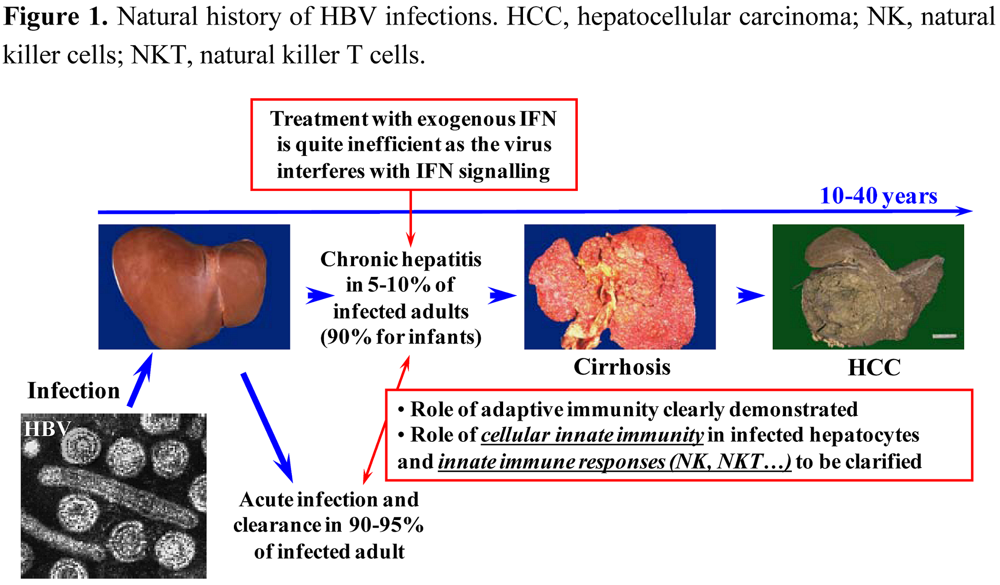
:1. Introduction


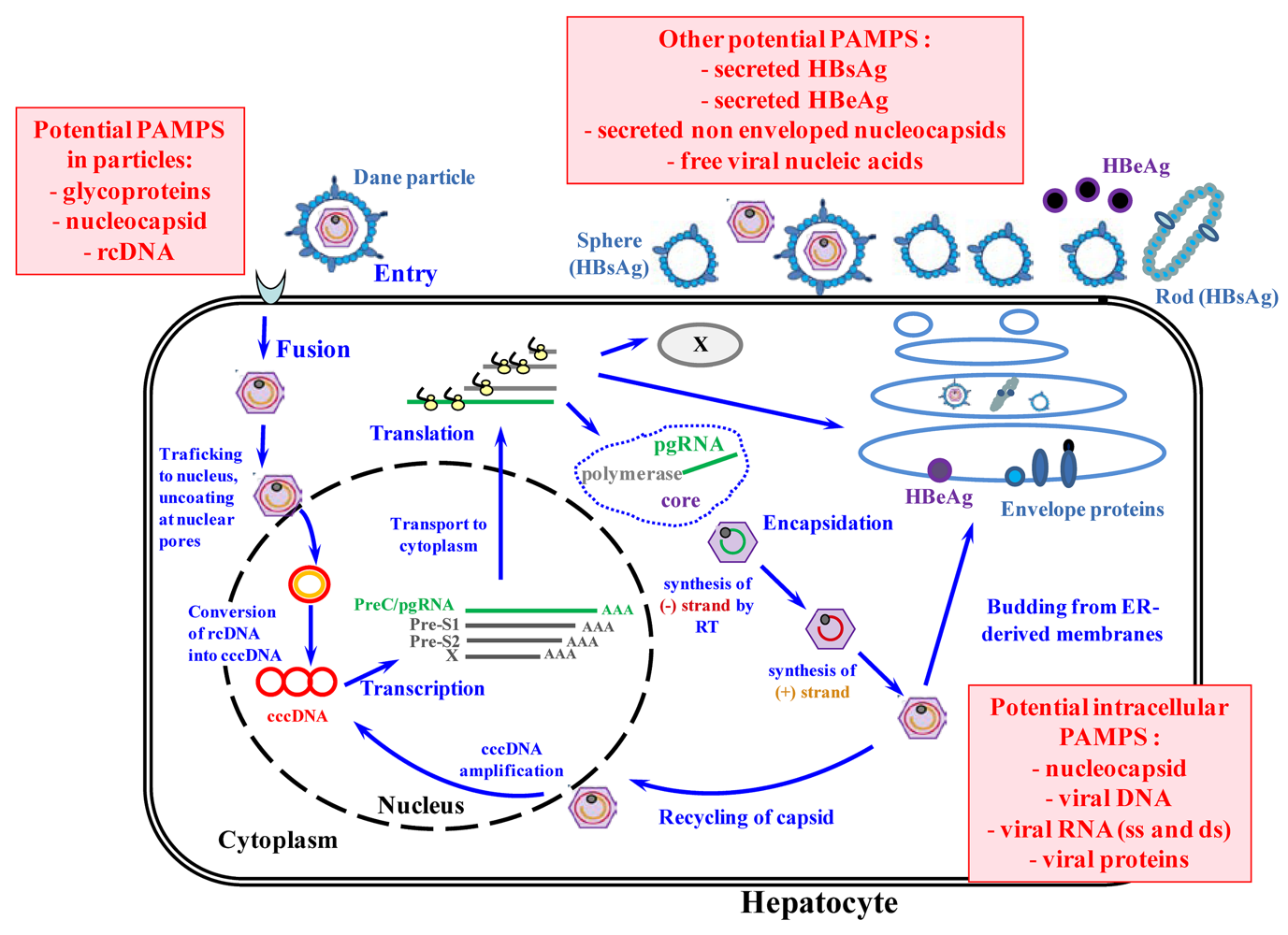
2. HBV, a stealth virus that does not elicit innate immunity?
3. Can HBV, as other viruses, be sensed by the immune system?

4. Can HBV inhibit type-I interferon pathways?
5. Can HBV inhibit host sensors of the innate response?

6. Concluding remarks
References
- Dienstag, J.L. Hepatitis B virus infection. N. Engl. J. Med. 2008, 359, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Lavanchy, D. Worldwide epidemiology of HBV infection, disease burden, and vaccine prevention . J. Clin. Virol. 2005, 34 (Suppl. 1), S1–S3. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Gehring, A.J. The immune response during hepatitis B virus infection. J. Gen. Virol. 2006, 87, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef]
- Biron, C.A.; Sen, G.N. Innate responses to viral infections. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; 2007; Lippincott Williams & Wilkins: Philadelphia, PA, USA. [Google Scholar]
- Gao, B.; Jeong, W.I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ . Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [PubMed]
- Liu, S.; Gallo, D.J.; Green, A.M.; Williams, D.L.; Gong, X.; Shapiro, R.A.; Gambotto, A.A.; Humphris, E.L.; Vodovotz, Y.; Billiar, T.R. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect. Immun. 2002, 70, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Naito, S. Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab. Pharmacokinet. 2005, 20, 452–477. [Google Scholar] [CrossRef] [PubMed]
- Ojaniemi, M.; Liljeroos, M.; Harju, K.; Sormunen, R.; Vuolteenaho, R.; Hallman, M. TLR-2 is upregulated and mobilized to the hepatocyte plasma membrane in the space of Disse and to the Kupffer cells TLR-4 dependently during acute endotoxemia in mice. Immunol. Lett. 2006, 102, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdorfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [PubMed]
- Vogel, S.N.; Fitzgerald, K.A.; Fenton, M.J. TLRs: differential adapter utilization by toll-like receptors mediates TLR-specific patterns of gene expression. Mol. Interv. 2003, 3, 466–477. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system . Science 327, 291–295. [CrossRef] [PubMed]
- Webster, G.J.; Reignat, S.; Maini, M.K.; Whalley, S.A.; Ogg, G.S.; King, A.; Brown, D.; Amlot, P.L.; Williams, R.; Vergani, D.; Dusheiko, G.M.; Bertoletti, A. Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms. Hepatology 2000, 32, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Menne, S.; Cote, P.J. The woodchuck as an animal model for pathogenesis and therapy of chronic hepatitis B virus infection. World J. Gastroenterol. 2007, 13, 104–124. [Google Scholar] [PubMed]
- Wieland, S.F.; Chisari, F.V. Stealth and cunning: hepatitis B and hepatitis C viruses. J. Virol. 2005, 79, 9369–9380. [Google Scholar] [CrossRef] [PubMed]
- Coffin, C.S.; Michalak, T.I. Persistence of infectious hepadnavirus in the offspring of woodchuck mothers recovered from viral hepatitis. J. Clin. Invest. 1999, 104, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Lew, Y.Y.; Michalak, T.I. In vitro and in vivo infectivity and pathogenicity of the lymphoid cell-derived woodchuck hepatitis virus. J. Virol. 2001, 75, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 6669–6674. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; Self, S.G.; Borrow, P. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; Dusheiko, G.M.; Jacobs, M.; Klenerman, P.; Maini, M.K. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.T.; Koh, S.; Goh, W.; Zhe, H.Y.; Gehring, A.J.; Lim, S.G.; Bertoletti, A. A longitudinal analysis of innate and adaptive immune profile during hepatic flares in chronic hepatitis B . J. Hepatol. 52, 330–339. [CrossRef] [PubMed]
- Guy, C.S.; Mulrooney-Cousins, P.M.; Churchill, N.D.; Michalak, T.I. Intrahepatic expression of genes affiliated with innate and adaptive immune responses immediately after invasion and during acute infection with woodchuck hepadnavirus. J. Virol. 2008, 82, 8579–8591. [Google Scholar] [CrossRef] [PubMed]
- McClary, H.; Koch, R.; Chisari, F.V.; Guidotti, L.G. Relative sensitivity of hepatitis B virus and other hepatotropic viruses to the antiviral effects of cytokines. J. Virol. 2000, 74, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Morris, A.; Mendez, H.; Koch, R.; Silverman, R.H.; Williams, B.R.; Chisari, F.V. Interferon-regulated pathways that control hepatitis B virus replication in transgenic mice. J. Virol. 2002, 76, 2617–2621. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lu, L.; Yuen, M.F.; Lam, T.W.; Chung, C.P.; Lam, C.L.; Zhang, B.; Wang, S.; Chen, Y.; Wu, S.H.; Poon, V.K.; Ng, F.; Chan, C.C.; Jiang, S.; Yuen, K.Y.; Zheng, B.J. Polymorphisms of type I interferon receptor 1 promoter and their effects on chronic hepatitis B virus infection. J. Hepatol. 2007, 46, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Smith, D.K.; Lu, L.; Poon, V.K.; Ng, F.; Chen, D.Q.; Huang, J.D.; Yuen, K.Y.; Cao, K.Y.; Zheng, B.J. A non-synonymous single nucleotide polymorphism in IFNAR1 affects susceptibility to chronic hepatitis B virus infection. J. Viral. Hepat. 2009, 16, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Diot, C.; Theze, N.; Fourel, I.; Loreal, O.; Brechot, C.; Guguen-Guillouzo, C. Hepatitis B virus infection of adult human hepatocytes cultured in the presence of dimethyl sulfoxide. J. Virol. 1988, 62, 4136–4143. [Google Scholar] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed]
- Hosel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; Langenkamp, A.; Falk, C.; Buning, H.; Rose-John, S.; Protzer, U. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Trautwein, C. Intracellular survival pathways in the liver. Liver Int. 2006, 26, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Gealy, C.; Denson, M.; Humphreys, C.; McSharry, B.; Wilkinson, G.; Caswell, R. Posttranscriptional suppression of interleukin-6 production by human cytomegalovirus. J. Virol. 2005, 79, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C. Kinetics of the immune response during HBV and HCV infection. Hepatology 2003, 38, 4–13. [Google Scholar] [CrossRef]
- Apostolou, E.; Thanos, D. Virus Infection Induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell 2008, 134, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Mossman, K.L.; Macgregor, P.F.; Rozmus, J.J.; Goryachev, A.B.; Edwards, A.M.; Smiley, J.R. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 2001, 75, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Christen, V.; Duong, F.; Bernsmeier, C.; Sun, D.; Nassal, M.; Heim, M.H. Inhibition of alpha interferon signaling by hepatitis B virus. J. Virol. 2007, 81, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Quiroga, J.A.; Carreno, V. Hepatitis B virus downregulates the human interferon-inducible MxA promoter through direct interaction of precore/core proteins. J. Gen. Virol. 2003, 84, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Rosmorduc, O.; Sirma, H.; Soussan, P.; Gordien, E.; Lebon, P.; Horisberger, M.; Brechot, C.; Kremsdorf, D. Inhibition of interferon-inducible MxA protein expression by hepatitis B virus capsid protein . J. Gen. Virol. 1999, 80 (Pt 5), 1253–1262. [Google Scholar] [PubMed]
- Lucifora, J.; Durantel, D.; Belloni, L.; Barraud, L.; Villet, S.; Vincent, I.E.; Margeridon-Thermet, S.; Hantz, O.; Kay, A.; Levrero, M.; Zoulim, F. Initiation of hepatitis B virus genome replication and production of infectious virus following delivery in HepG2 cells by novel recombinant baculovirus vector. J. Gen. Virol. 2008, 89, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Durantel, D.; Testoni, B.; Hantz, O.; Levrero, M.; Zoulim, F. Control of hepatitis B virus replication by innate response of HepaRG cells . Hepatology 2009. [Google Scholar]
- Fisicaro, P.; Valdatta, C.; Boni, C.; Massari, M.; Mori, C.; Zerbini, A.; Orlandini, A.; Sacchelli, L.; Missale, G.; Ferrari, C. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut 2009, 58, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Twu, J.S.; Lee, C.H.; Lin, P.M.; Schloemer, R.H. Hepatitis B virus suppresses expression of human beta-interferon. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Whitten, T.M.; Quets, A.T.; Schloemer, R.H. Identification of the hepatitis B virus factor that inhibits expression of the beta interferon gene. J. Virol. 1991, 65, 4699–4704. [Google Scholar] [PubMed]
- Guan, S.H.; Lu, M.; Grunewald, P.; Roggendorf, M.; Gerken, G.; Schlaak, J.F. Interferon-alpha response in chronic hepatitis B-transfected HepG2 2.15 cells is partially restored by lamivudine treatment. World J. Gastroenterol. 2007, 13, 228–235. [Google Scholar] [PubMed]
- Fernandez, M.; Quiroga, J.A.; Martin, J.; Cotonat, T.; Pardo, M.; Horisberger, M.A.; Carreno, V. Impaired interferon induction of human MxA protein in chronic hepatitis B virus infection. J. Med. Virol. 1997, 51, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Rosmorduc, O.; Petit, M.A.; Pol, S.; Capel, F.; Bortolotti, F.; Berthelot, P.; Brechot, C.; Kremsdorf, D. In vivo and in vitro expression of defective hepatitis B virus particles generated by spliced hepatitis B virus RNA. Hepatology 1995, 22, 10–19. [Google Scholar] [PubMed]
- Soussan, P.; Pol, J.; Garreau, F.; Schneider, V.; Pendeven, C.L.; Nalpas, B.; Lacombe, K.; Bonnard, P.; Pol, S.; Kremsdorf, D. Expression of Defective Hepatitis B Virus Particles Derived from Singly Spliced RNA Is Related to Liver Disease . J. Infect. Dis. 2008. [Google Scholar]
- Foster, G.R.; Ackrill, A.M.; Goldin, R.D.; Kerr, I.M.; Thomas, H.C.; Stark, G.R. Expression of the terminal protein region of hepatitis B virus inhibits cellular responses to interferons alpha and gamma and double-stranded RNA. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 2888–2892. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.R.; Goldin, R.D.; Hay, A.; McGarvey, M.J.; Stark, G.R.; Thomas, H.C. Expression of the terminal protein of hepatitis B virus is associated with failure to respond to interferon therapy. Hepatology 1993, 17, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.R.; Ackrill, A.M.; Goldin, R.D.; Kerr, I.M.; Thomas, H.C.; Stark, G.R. Corections : expression of the terminal protein region of hepatitis B virus inhibits cellular responses to interferons alpha and gamma and double-stranded RNA. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 3632. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Xu, Y.; Lin, S.; Zhang, X.; Xiang, L.; Yuan, Z. Hepatitis B virus polymerase inhibits the interferon-inducible MyD88 promoter by blocking nuclear translocation of Stat1. J. Gen. Virol. 2007, 88, 3260–3269. [Google Scholar] [CrossRef] [PubMed]
- Duong, F.H.; Filipowicz, M.; Tripodi, M.; La Monica, N.; Heim, M.H. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology 2004, 126, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Imanishi, H.; Morisaki, H.; Liu, W.; Nakamura, H.; Morisaki, T.; Hada, T. Recombinant HBsAg inhibits LPS-induced COX-2 expression and IL-18 production by interfering with the NFkappaB pathway in a human monocytic cell line, THP-1. J. Hepatol. 2005, 43, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.M. Viral recognition by Toll-like receptors. Semin. Immunol. 2007, 19, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, K.; Skinner, N.A.; Thompson, A.J.; Riordan, S.M.; Sozzi, V.; Edwards, R.; Rodgers, S.; Kurtovic, J.; Chang, J.; Lewin, S.; Desmond, P.; Locarnini, S. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 2007, 45, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Isogawa, M.; Robek, M.D.; Furuichi, Y.; Chisari, F.V. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 2005, 79, 7269–7272. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cheng, Y.; Xu, Y.; Liao, J.; Zhang, X.; Hu, Y.; Zhang, Q.; Wang, J.; Zhang, Z.; Shen, F.; Yuan, Z. Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin. Immunol. 2008, 128, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Meng, Z.; Jiang, M.; Pei, R.; Trippler, M.; Broering, R.; Bucchi, A.; Sowa, J.P.; Dittmer, U.; Yang, D.; Roggendorf, M.; Gerken, G.; Lu, M.; Schlaak, J.F. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 2009, 49, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald-Bocarsly, P.; Dai, J.; Singh, S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008, 19, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Shen, H.C.; Jia, N.N.; Wang, H.; Lin, L.Y.; An, B.Y.; Gui, H.L.; Guo, S.M.; Cai, W.; Yu, H.; Guo, Q.; Bao, S. Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9 . Microbes Infect. 2009. [Google Scholar]
- Li, N.; Li, Q.; Qian, Z.; Zhang, Y.; Chen, M.; Shi, G. Impaired TLR3/IFN-beta signaling in monocyte-derived dendritic cells from patients with acute-on-chronic hepatitis B liver failure: relevance to the severity of liver damage. Biochem. Biophys. Res. Commun. 2009, 390, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli, S.; Schwerin, W.; Rohwer, A.; Hoffmann, S.; Weyer, S.; Weth, R.; Meisel, H.; Diepolder, H.; Geissler, M.; Galle, P.R.; Lohr, H.F.; Bocher, W.O. Phenotype and function of monocyte derived dendritic cells in chronic hepatitis B virus infection. J. Gen. Virol. 2004, 85, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.S.; Xing, L.H.; Liu, M.X.; Zhu, C.L.; Liu, H.G.; Wang, H.F.; Lei, Z.Y. Dysfunction of peripheral blood dendritic cells from patients with chronic hepatitis B virus infection. World J. Gastroenterol. 2001, 7, 537–541. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Ait-goughoulte, M.; Lucifora, J.; Zoulim, F.; Durantel, D. Innate Antiviral Immune Responses to Hepatitis B Virus. Viruses 2010, 2, 1394-1410. https://doi.org/10.3390/v2071394
Ait-goughoulte M, Lucifora J, Zoulim F, Durantel D. Innate Antiviral Immune Responses to Hepatitis B Virus. Viruses. 2010; 2(7):1394-1410. https://doi.org/10.3390/v2071394
Chicago/Turabian StyleAit-goughoulte, Malika, Julie Lucifora, Fabien Zoulim, and David Durantel. 2010. "Innate Antiviral Immune Responses to Hepatitis B Virus" Viruses 2, no. 7: 1394-1410. https://doi.org/10.3390/v2071394
APA StyleAit-goughoulte, M., Lucifora, J., Zoulim, F., & Durantel, D. (2010). Innate Antiviral Immune Responses to Hepatitis B Virus. Viruses, 2(7), 1394-1410. https://doi.org/10.3390/v2071394