JaPaFi: A Novel Program for the Identification of Highly Conserved DNA Sequences


Abstract
:
1. Introduction
2. Materials and Methods
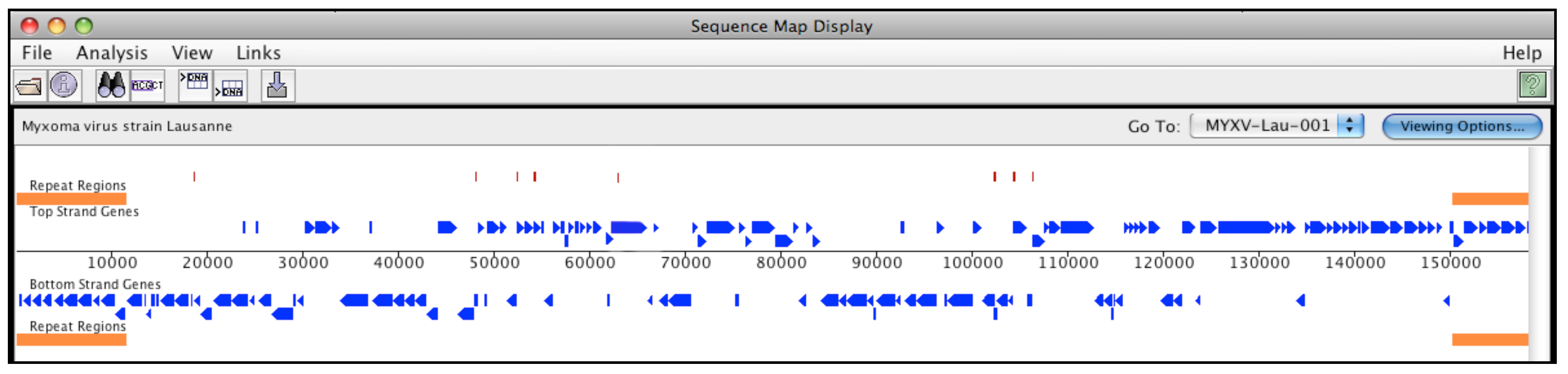
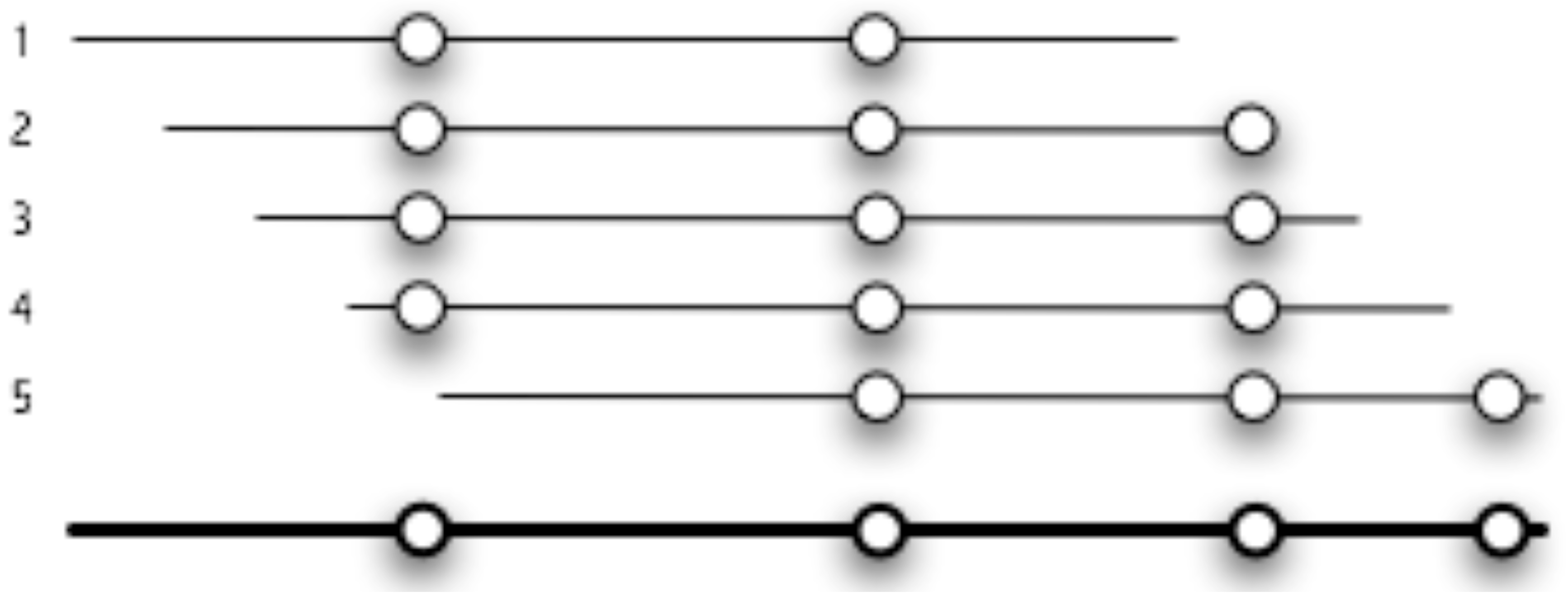
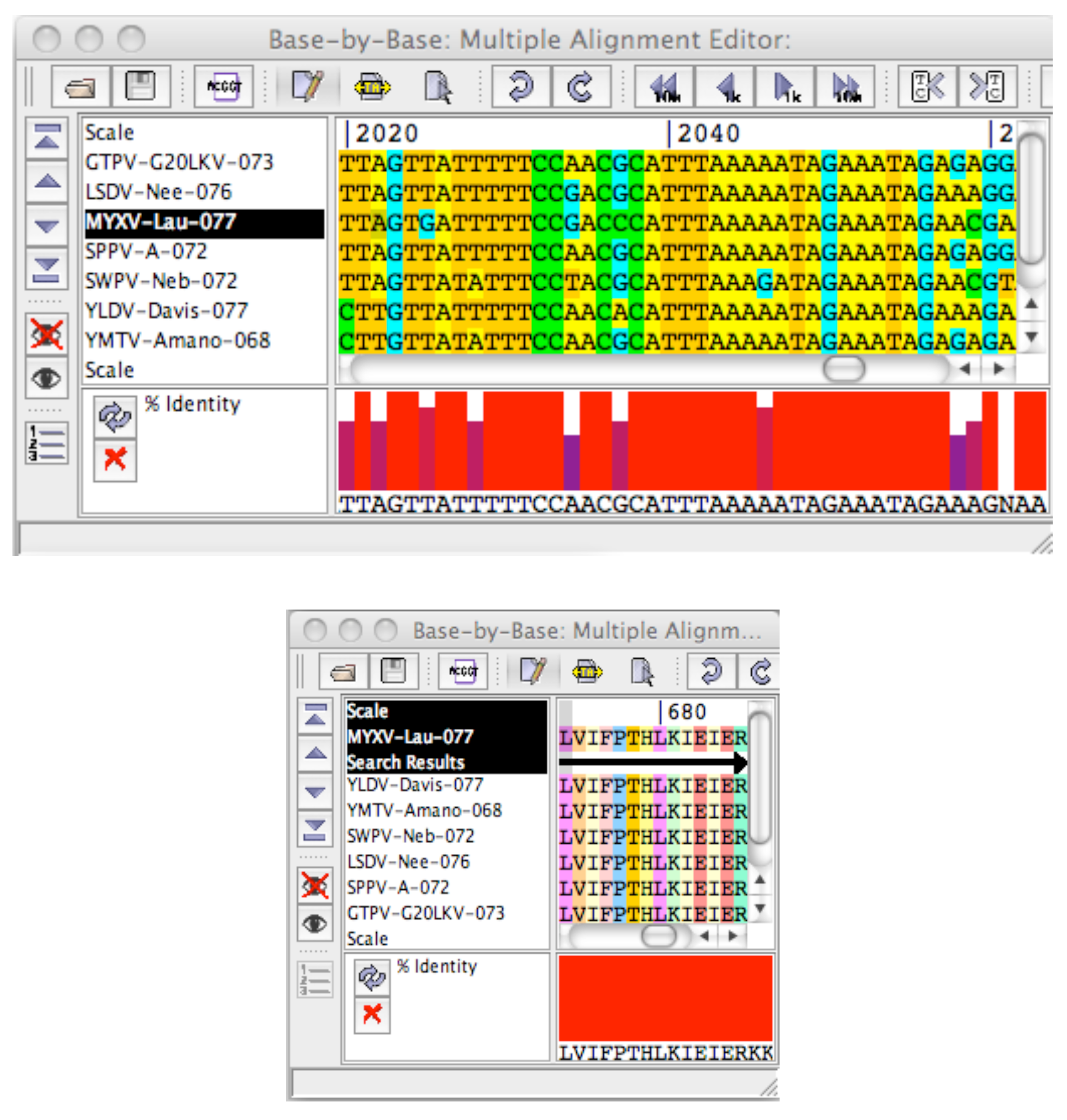
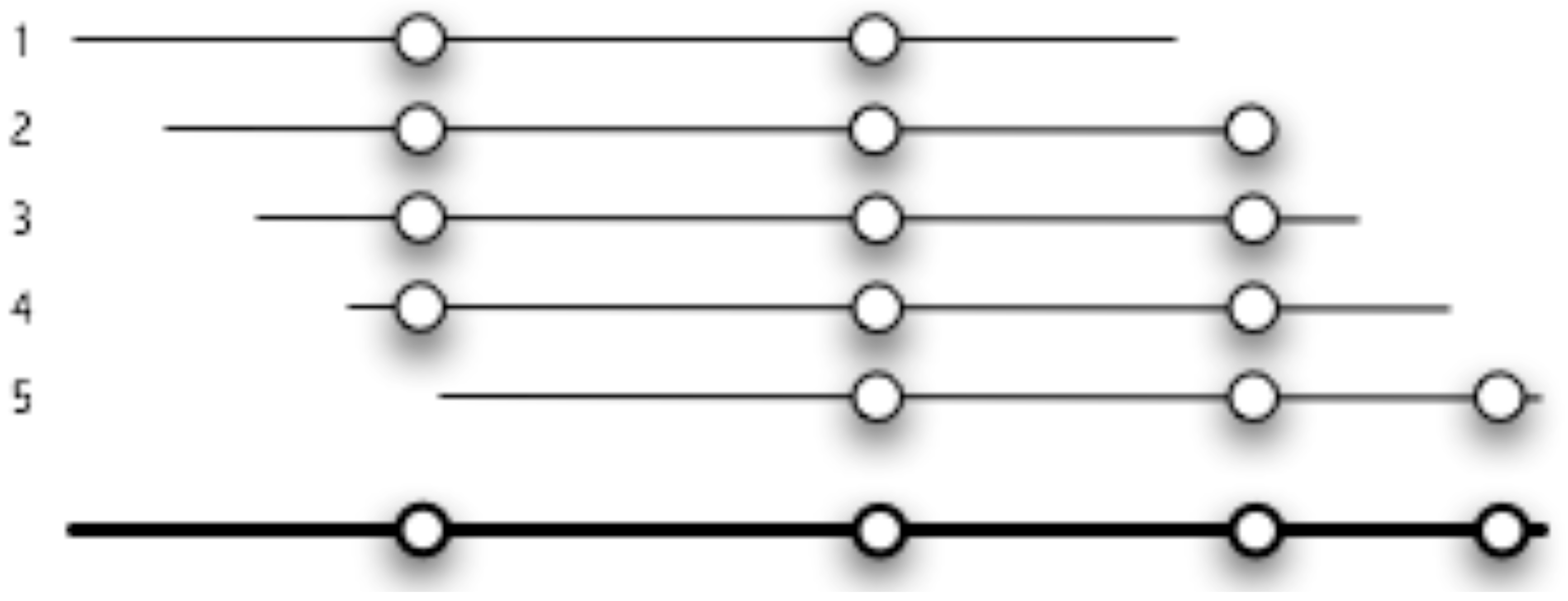
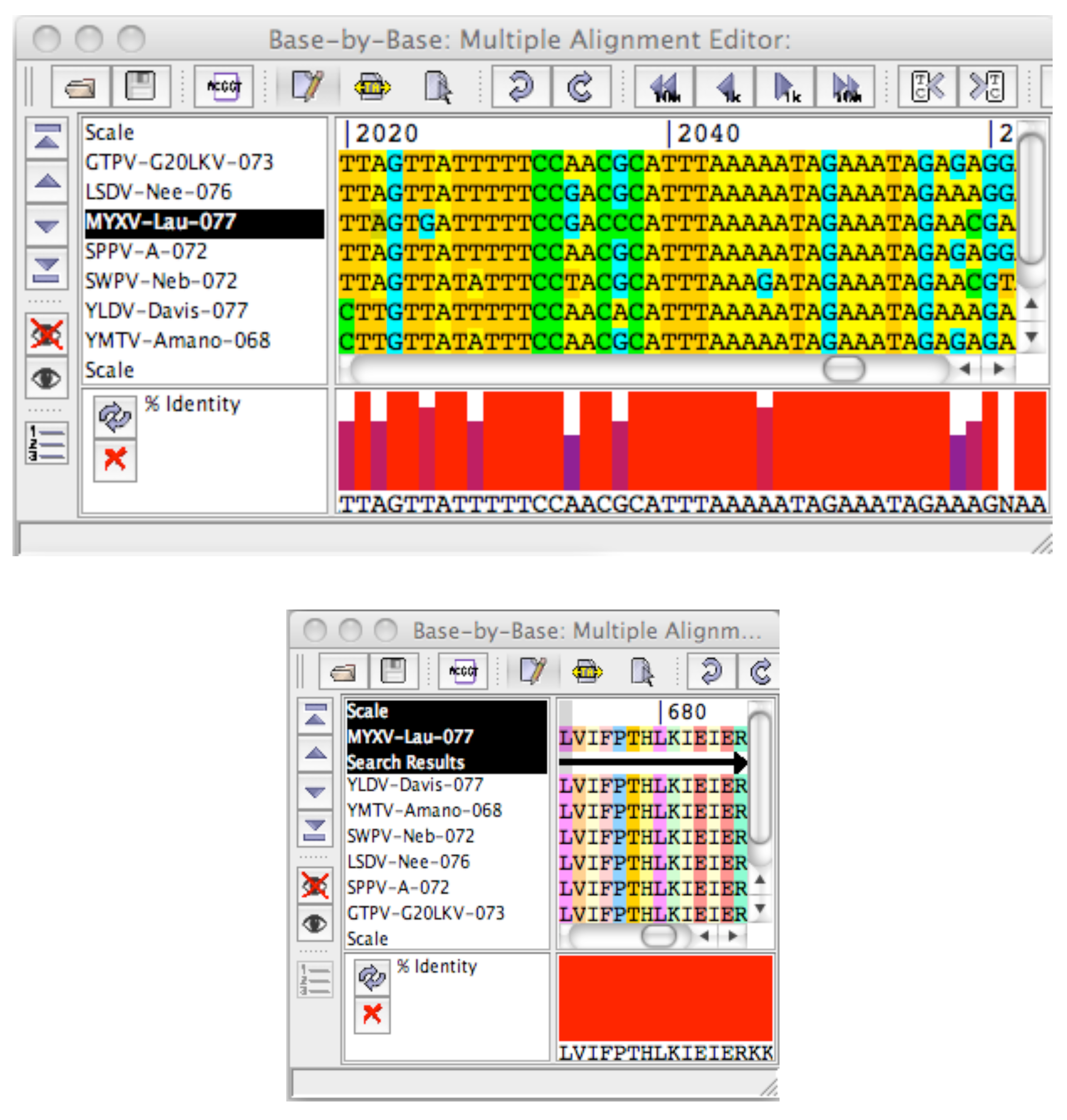
2.1. Description of Java Pattern Finder (JaPaFi)
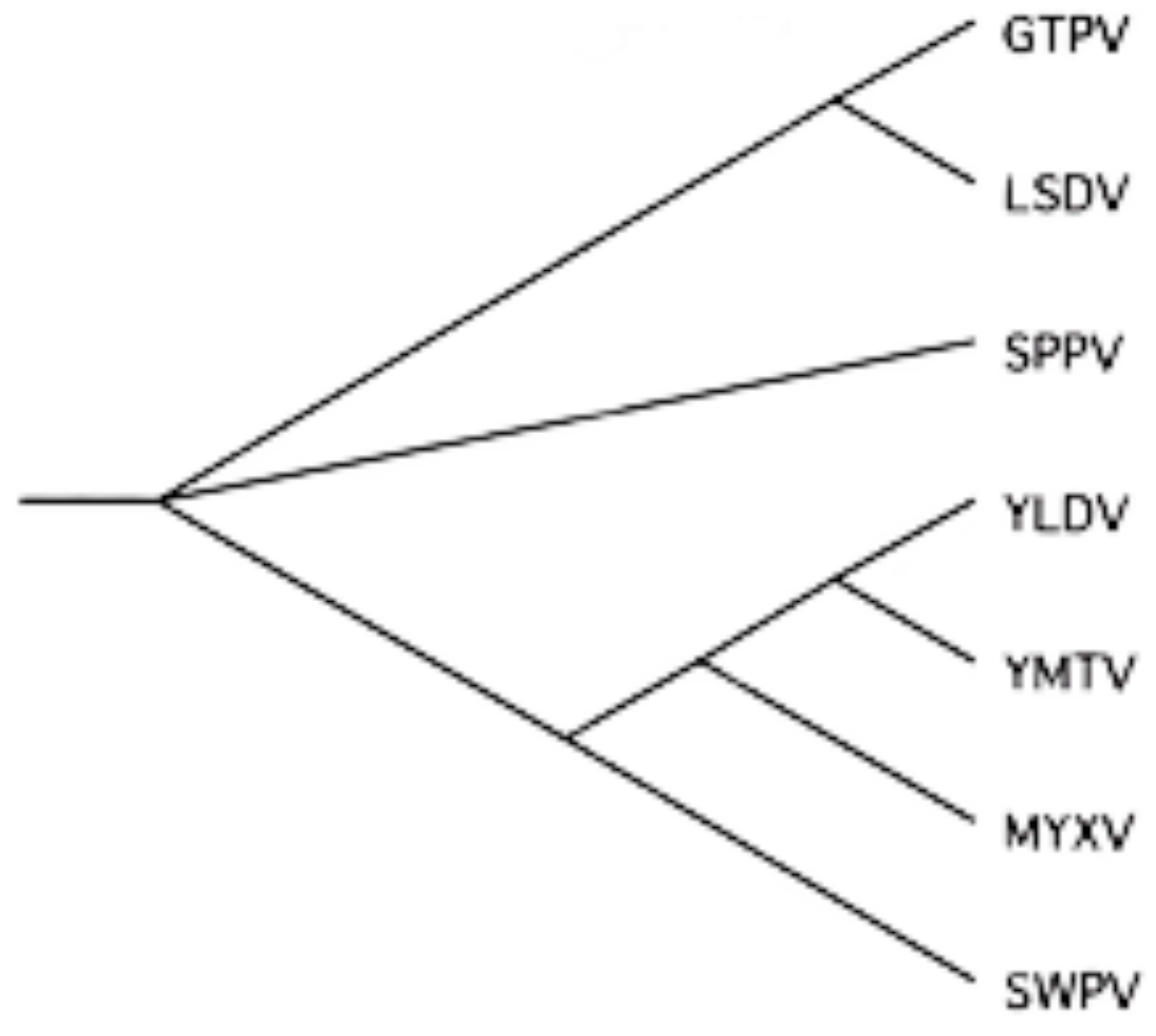
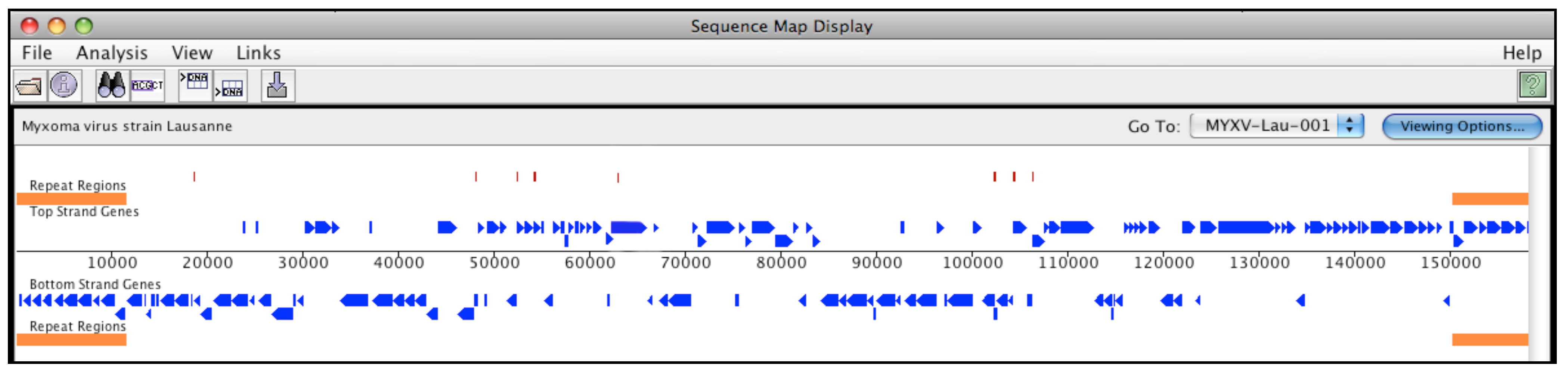
2.2. Genomes included in this study
3. Results and Discussion
3.1. Counting the number of hits as the parameters of length and number of differences are varied
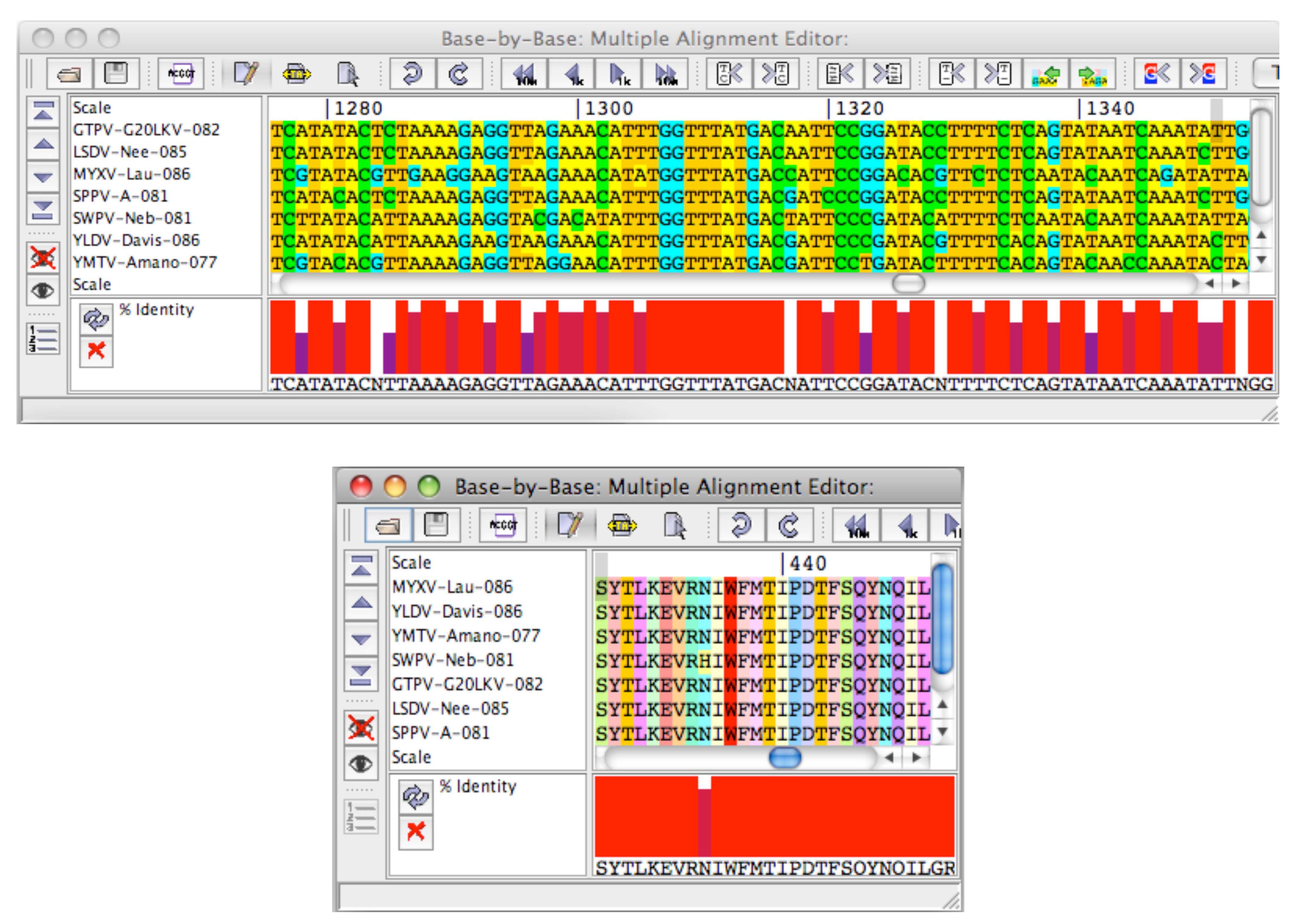
3.2. Quantifying the degree of conservation
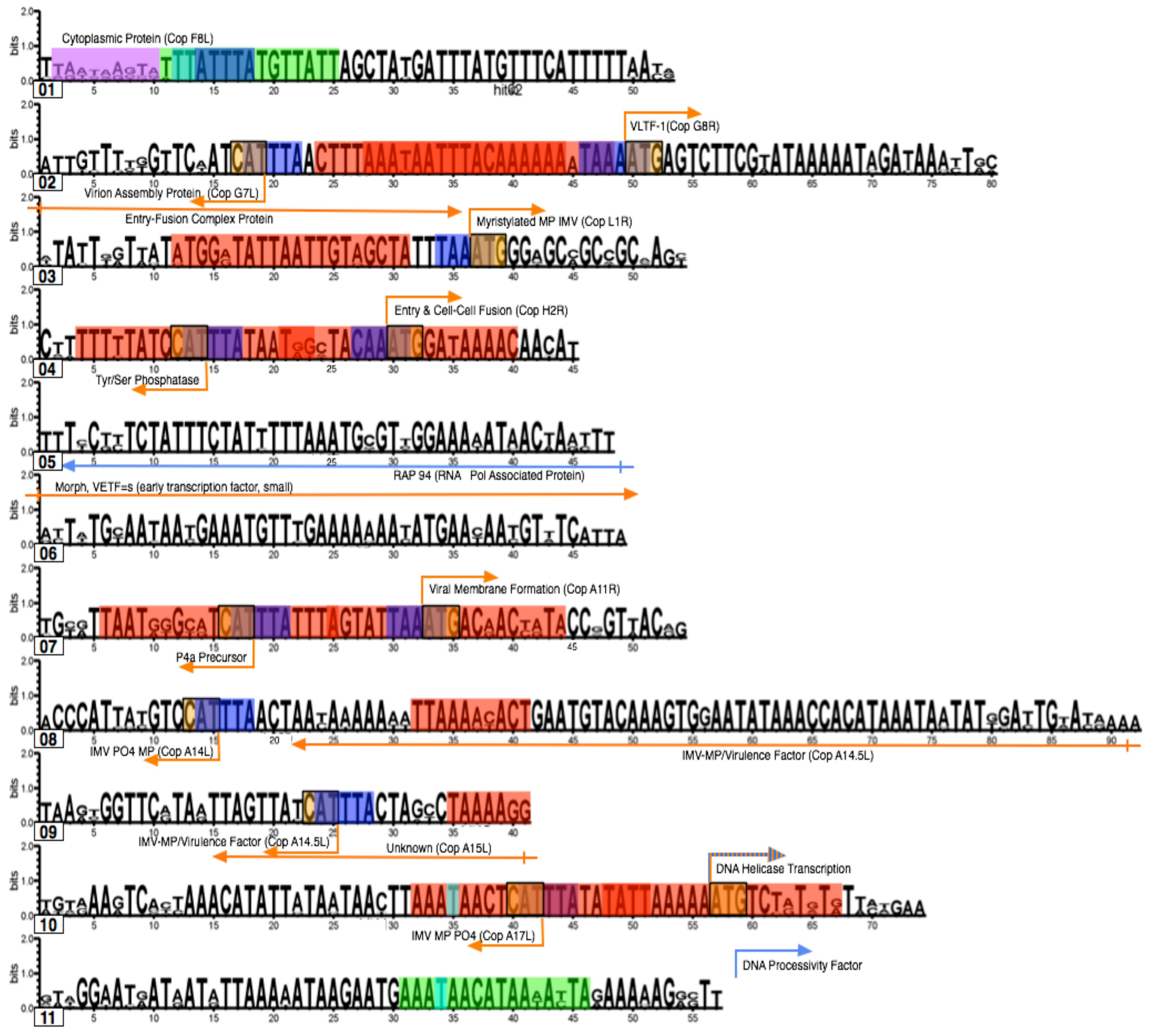
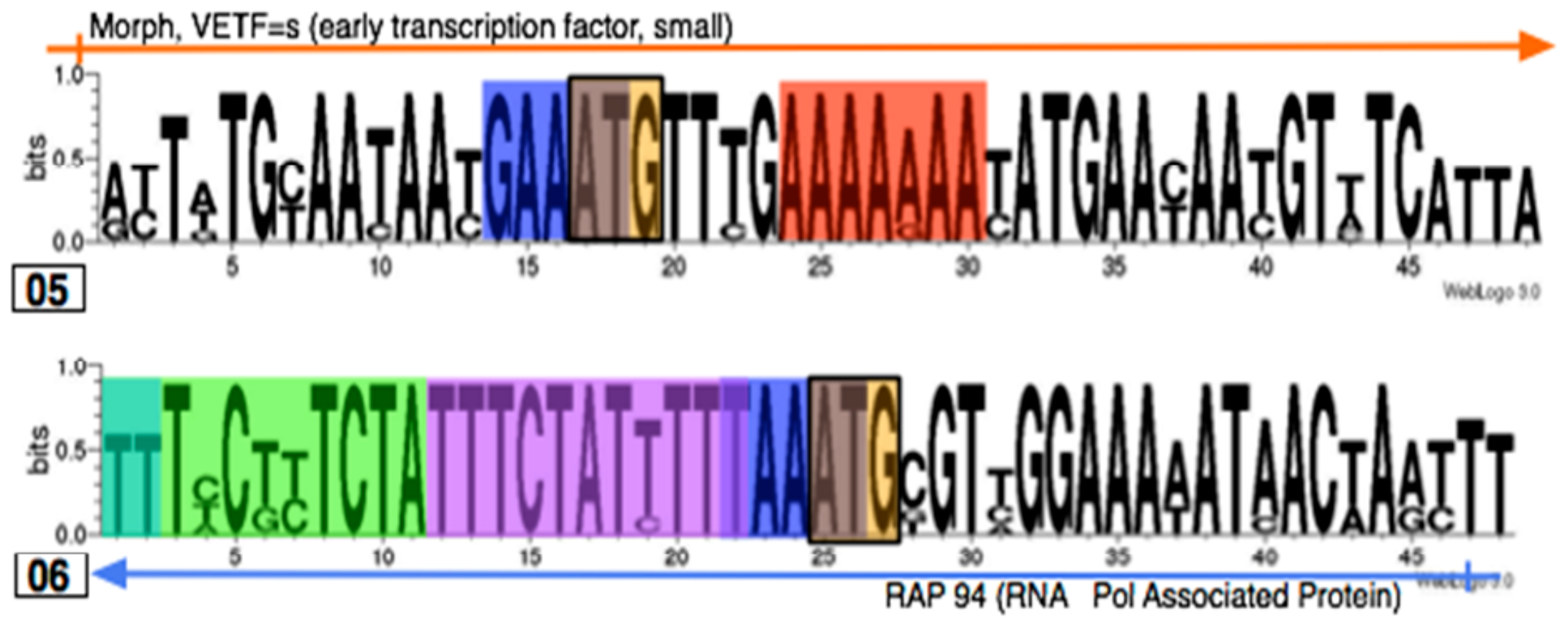
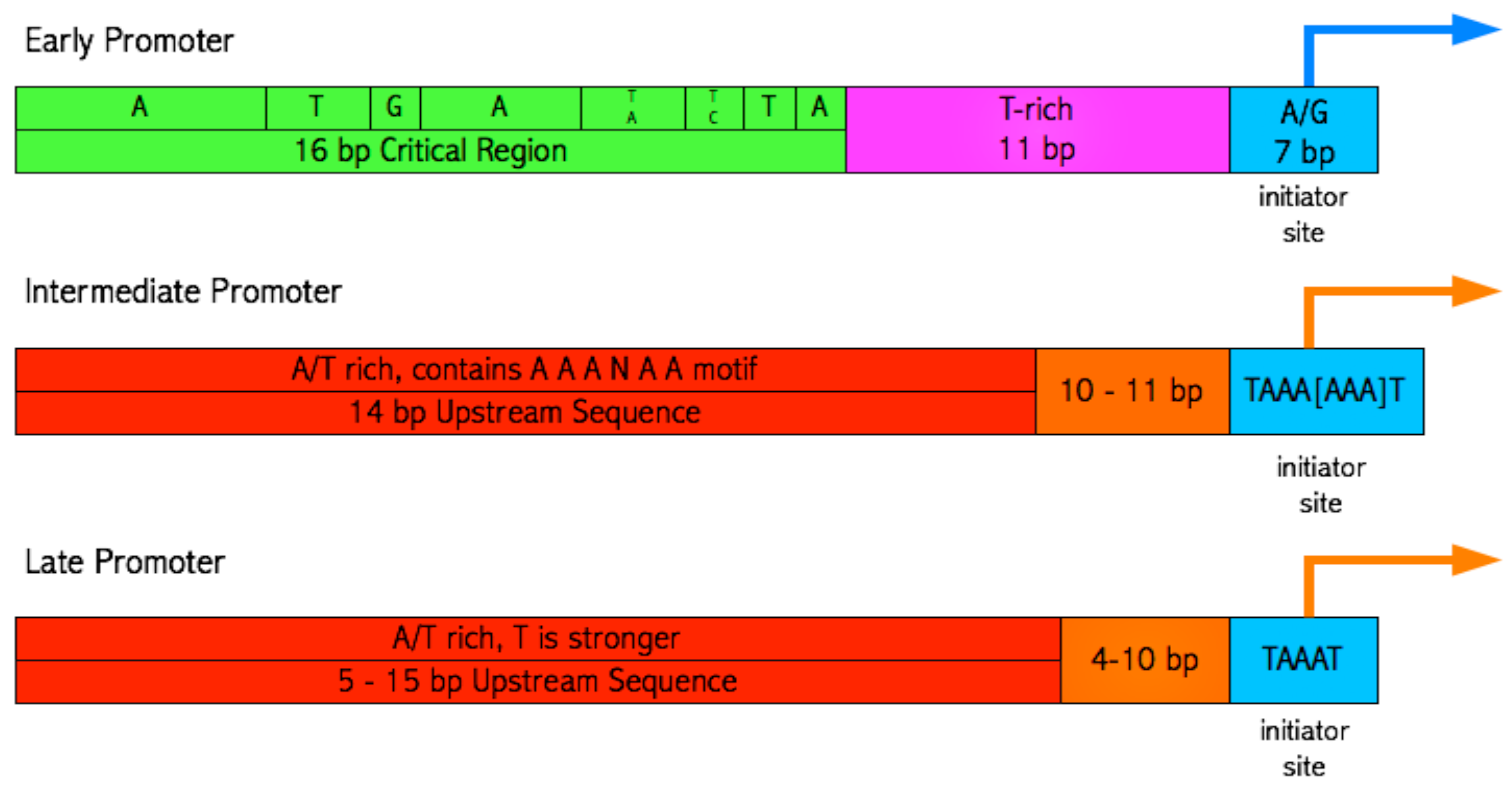
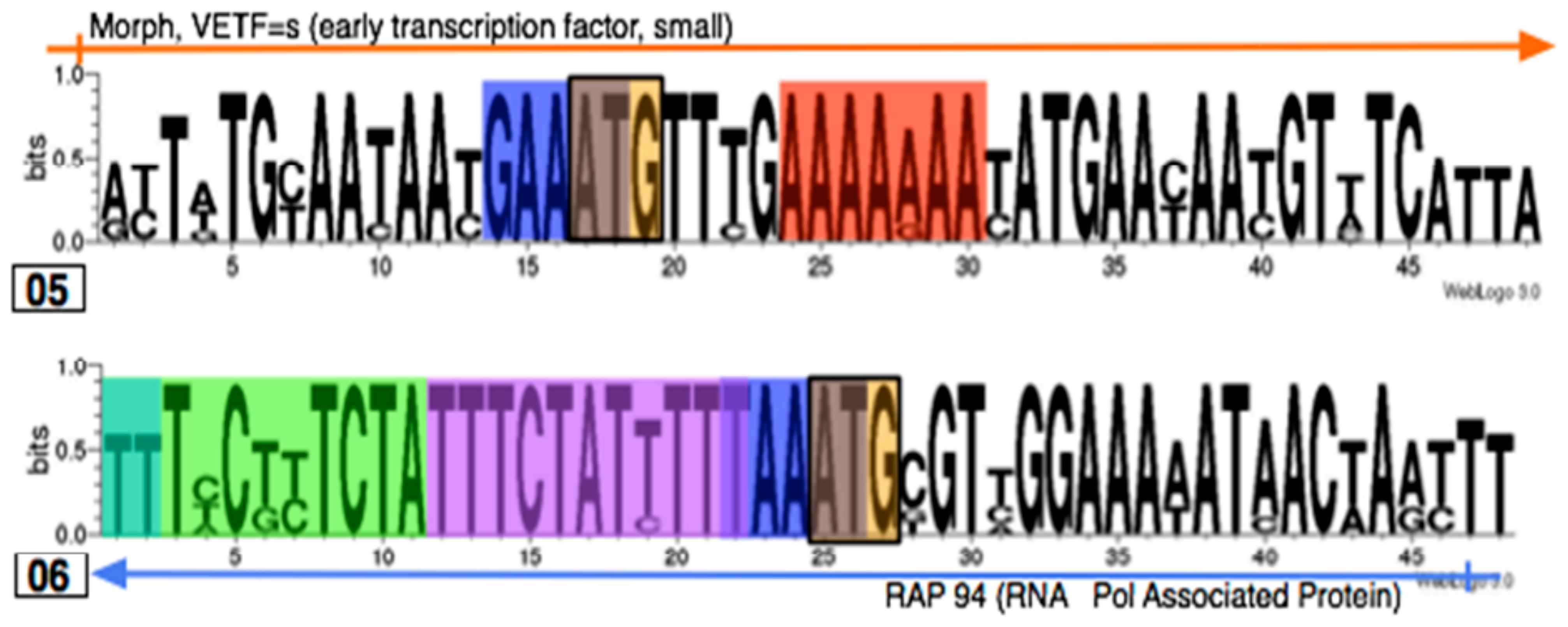
3.3. Identifying promoter elements within the hits
3.4. Searching for short motifs shared between the hits and early, intermediate and late promoters
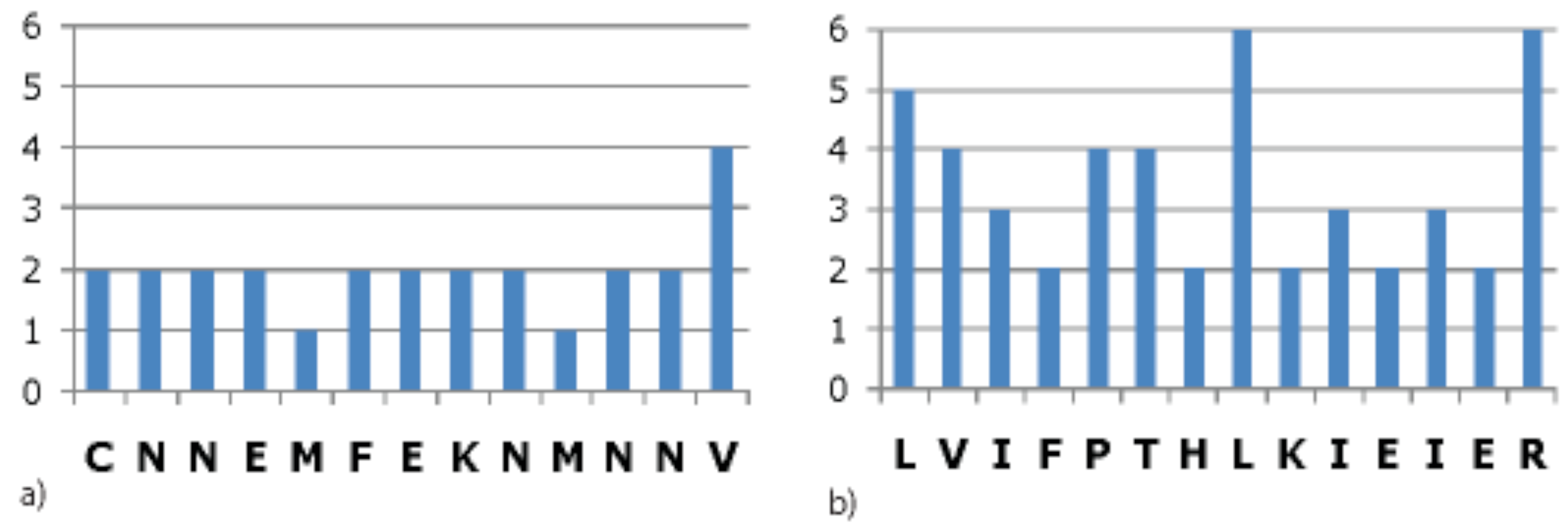
3.5. Coding Region Hits
3.5.1. Conserved Protein Domains
3.5.2. Codon Degeneracy
4. Conclusions
- Future Work
Acknowledgments
References and Notes
- Hardison, R.C. Comparative genomics. PLoS Biol. 2003, 1, E58. [Google Scholar] [CrossRef] [PubMed]
- Sivashankari, S.; Shanmughavel, P. Comparative genomics - a perspective. Bioinformation 2007, 1, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Kellis, M.; Patterson, N.; Endrizzi, M.; Birren, B.; Lander, E.S. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 2003, 423, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Zheng, W.J. Combining comparative genomics with de novo motif discovery to identify human transcription factor DNA-binding motifs. BMC Bioinformatics 2006, 7 (Suppl. 4), S21. [Google Scholar] [CrossRef]
- Cui, X.; Vinar, T.; Brejova, B.; Shasha, D.; Li, M. Homology search for genes. Bioinformatics 2007, 23, i97–i103. [Google Scholar] [CrossRef]
- She, R.; Chu, J.S.; Wang, K.; Pei, J.; Chen, N. GenBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res. 2009, 19, 143–149. [Google Scholar] [CrossRef]
- Seneff, S.; Wang, C.; Burge, C.B. Gene structure prediction using an orthologous gene of known exon-intron structure. Appl. Bioinformatics 2004, 3, 81–90. [Google Scholar] [CrossRef]
- Da Silva, M.; Upton, C. Vaccinia virus G8R protein: A structural ortholog of proliferating cell nuclear antigen (PCNA). PLoS One 2009, 4, e5479. [Google Scholar] [CrossRef]
- Brunetti, C.R.; Amano, H.; Ueda, Y.; Qin, J.; Miyamura, T.; Suzuki, T.; Li, X.; Barrett, J.W.; McFadden, G. Complete genomic sequence and comparative analysis of the tumorigenic poxvirus Yaba monkey tumor virus. J. Virol. 2003, 77, 13335–13347. [Google Scholar] [CrossRef]
- Eaton, H.E.; Metcalf, J.; Brunetti, C.R. Characterization of the promoter activity of a poxvirus conserved element. Can. J. Microbiol. 2008, 54, 483–488. [Google Scholar] [CrossRef]
- Coupar, B.E.; Boyle, D.B.; Both, G.W. Effect of in vitro mutations in a vaccinia virus early promoter region monitored by herpes simplex virus thymidinekinase expression in recombinant vaccinia virus. J. Gen. Virol. 1987, 68 Pt 9, 2299–2309. [Google Scholar] [CrossRef]
- Fick, W.C.; Viljoen, G.J. Identification and characterisation of an early/late bi-directional promoter of the capripoxvirus, lumpy skin disease virus. Arch. Virol. 1999, 144, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Moss, B. Structure of vaccinia virus early promoters. J. Mol. Biol. 1989, 210, 749–769. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Moss, B. Structure of vaccinia virus late promoters. J. Mol. Biol. 1989, 210, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Baillie, D.L.; Rose, A.M. WABA success: A tool for sequence comparison between large genomes. Genome Res. 2000, 10, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Vmatch Home Page. http://www.vmatch.de/ (accessed on 3 August 2010).
- Hohl, M.; Kurtz, S.; Ohlebusch, E. Efficient multiple genome alignment. Bioinformatics 2002, 18 (Suppl. 1), S312–S320. [Google Scholar] [CrossRef]
- Chain, P.; Kurtz, S.; Ohlebusch, E.; Slezak, T. An applications-focused review of comparative genomics tools: Capabilities, limitations and future challenges. Brief Bioinform 2003, 4, 105–123. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Baeza-Yates, R.A.; Gonnet, G.H. A Fast Algorithm on Average for All-Against-All Sequence Matching. In Proceedings of the String Processing and information Retrieval Symposium & international Workshop on Groupware, SPIRE. IEEE Computer Society, Washington, DC, USA, September 1999; pp. 16–23. [Google Scholar]
- Barsky, M.S.U.; Thomo, A.; Upton, C. A graph approach to the threshold all-against-all substring matching problem. JEA 2008, 12. [Google Scholar] [CrossRef]
- Viral Bioinformatics Resource Home Page. www.virology.ca (accessed on 3 August 2010).
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Moss, B. Interaction of the vaccinia virus RNA polymerase-associated 94-kilodalton protein with the early transcription factor. J. Virol. 2009, 83, 12018–12026. [Google Scholar] [CrossRef] [PubMed]
- Baldick, C.J., Jr.; Keck, J.G.; Moss, B. Mutational analysis of the core, spacer, and initiator regions of vaccinia virus intermediate-class promoters. J. Virol. 1992, 66, 4710–4719. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.; Ahn, B.Y.; Amegadzie, B.; Gershon, P.D.; Keck, J.G. Cytoplasmic transcription system encoded by vaccinia virus. J. Biol. Chem. 1991, 266, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- BLAST tutorial Home Page. http://www.ncbi.nlm.nih.gov/Education (accessed on 3 August 2010).
- Olafsdottir, G.; Svansson, V.; Ingvarsson, S.; Marti, E.; Torsteinsdottir, S. In vitro analysis of expression vectors for DNA vaccination of horses: The effect of a Kozak sequence. Acta Vet. Scand. 2008, 50, 44. [Google Scholar] [CrossRef]
- Sánchez-Puig, J.M.B.R. AUG context and mRNA translation in vaccinia virus. Span. J. Agric. Res. 2008, 6, 73–80. [Google Scholar] [CrossRef]
- Li, J.; Broyles, S.S. Recruitment of vaccinia virus RNA polymerase to an early gene promoter by the viral early transcription factor. J. Biol. Chem. 1993, 268, 2773–2780. [Google Scholar] [CrossRef]
- Ahn, B.Y.; Gershon, P.D.; Moss, B. RNA polymerase-associated protein Rap94 confers promoter specificity for initiating transcription of vaccinia virus early stage genes. J. Biol. Chem. 1994, 269, 7552–7557. [Google Scholar] [CrossRef]
- Condit, R.C. Vaccinia, Inc.--probing the functional substructure of poxviral replication factories. Cell Host Microbe 2007, 2, 205–207. [Google Scholar] [CrossRef]
- Zhou, H.; Jackson, A.O. Expression of the barley stripe mosaic virus RNA beta "triple gene block". Virology 1996, 216, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Juszczuk, M.; Zagorski-Ostoja, W.; Hulanicka, D.M. Studies on the translation mechanism of subgenomic RNA of potato leafroll virus. Acta Biochim. Pol. 1997, 44, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Upton, C. Screening predicted coding regions in poxvirus genomes. Virus Genes 2000, 20, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Marcheschi, R.J.; Staple, D.W.; Butcher, S.E. Programmed ribosomal frameshifting in SIV is induced by a highly structured RNA stem-loop. J. Mol. Biol. 2007, 373, 652–663. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Species | GenBank Accession | Abbreviation |
|---|---|---|---|
| Capripoxvirus | Goatpox virus strain G20-LKV | AY077836 | GTPV |
| Capripoxvirus | Lumpy skin disease virus strain Neethling 2490 | NC_003027 | LSDV |
| Leporipoxvirus | Myxoma virus strain Lausanne | NC_001132 | MYXV |
| Capripoxvirus | Sheeppox virus strain A | AY077833 | SPPV |
| Suipoxvirus | Swinepox virus strain Nebraska 17077-99 | NC_003389 | SWPV |
| Yatapoxvirus | Yaba-like disease virus strain Davis | NC_005179 | YLDV |
| Yatapoxvirus | Yaba monkey tumor virus strain Amano | NC_002632 | YMTV |
| S\K | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|---|
| 15 | 16 | 303 | ||||||
| 16 | 12 | 115 | ||||||
| 17 | 11 | 57 | ||||||
| 18 | 10 | 31 | 417 | |||||
| 19 | 9 | 27 | 189 | |||||
| 20 | 6 | 21 | 117 | |||||
| 21 | 5 | 15 | 70 | 423 | ||||
| 22 | 4 | 15 | 55 | 250 | ||||
| 23 | 3 | 13 | 47 | 177 | ||||
| 24 | 2 | 11 | 28 | 111 | ||||
| 25 | 2 | 11 | 25 | 98 | ||||
| 26 | 1 | 10 | 22 | 83 | ||||
| 27 | 1 | 8 | 15 | 50 | 148 | 464 | ||
| 28 | 1 | 7 | 15 | 45 | 130 | 358 | ||
| 29 | 1 | 5 | 13 | 37 | 284 | |||
| 30 | 1 | 4 | 9 | 24 | 76 | 188 | ||
| 31 | 1 | 4 | 6 | 24 | 65 | |||
| 32 | 1 | 3 | 6 | 20 | 60 | 148 | ||
| 33 | 0 | 3 | 5 | 14 | 34 | |||
| 34 | 0 | 3 | 5 | 12 | 30 | 93 | ||
| 35 | 0 | 3 | 4 | 10 | 27 | 184 | ||
| 36 | 0 | 3 | 4 | 9 | 22 | 61 | ||
| 37 | 0 | 3 | 4 | 8 | 19 | 115 | ||
| 38 | 0 | 3 | 4 | 8 | 14 | 43 | ||
| 39 | 0 | 2 | 4 | 4 | 11 | 80 | ||
| 40 | 0 | 2 | 4 | 3 | 10 | 28 | ||
| 41 | 0 | 2 | 3 | 3 | 9 | 26 | ||
| 42 | 0 | 1 | 3 | 3 | 6 | 16 | 47 | |
| 43 | 0 | 1 | 3 | 3 | 6 | 14 | 38 | |
| 44 | 0 | 1 | 3 | 3 | 6 | 12 | 35 | |
| 45 | 0 | 1 | 2 | 3 | 4 | 6 | 26 | |
| 46 | 0 | 1 | 2 | 3 | 3 | 5 | 25 | |
| 47 | 0 | 1 | 2 | 3 | 3 | 5 | 23 | |
| 48 | 0 | 1 | 2 | 3 | 3 | 5 | 18 | |
| 49 | 0 | 1 | 2 | 3 | 3 | 5 | 14 | |
| 50 | 0 | 1 | 2 | 3 | 3 | 4 | 12 | |
| 51 | 0 | 0 | 2 | 3 | 3 | 3 | 5 | |
| 52 | 0 | 0 | 2 | 3 | 3 | 3 | 5 | 18 |
| 53 | 0 | 0 | 1 | 3 | 3 | 3 | 5 | 18 |
| 54 | 0 | 0 | 1 | 3 | 3 | 3 | 4 | 11 |
| 55 | 0 | 0 | 1 | 2 | 3 | 3 | 3 | 9 |
| 56 | 0 | 0 | 1 | 1 | 2 | 3 | 3 | 8 |
| 57 | 0 | 0 | 1 | 1 | 2 | 3 | 3 | 5 |
| 58 | 0 | 0 | 0 | 1 | 2 | 3 | 3 | 5 |
| 59 | 0 | 0 | 0 | 1 | 1 | 3 | 3 | 5 |
| 60 | 0 | 0 | 0 | 1 | 1 | 3 | 3 | 5 |
| 61 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 |
| 62 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 |
| 63 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 |
| 64 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 |
| 65 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 |
| 66 | 0 | 0 | 0 | 0 | 1 | 2 | 3 | 3 |
| 67 | 0 | 0 | 0 | 0 | 0 | 2 | 3 | 3 |
| 68 | 0 | 0 | 0 | 0 | 0 | 2 | 3 | 3 |
| 69 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 3 |
| 70 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 3 |
| Final set of hits | |||
|---|---|---|---|
| hit | start | stop | length |
| 01 | 18521 | 18573 | 53 |
| 02 | 52335 | 52414 | 80 |
| 03 | 54146 | 54199 | 54 |
| 04 | 66601 | 66645 | 45 |
| 05 | 80165 | 80212 | 48 |
| 06 | 68525 | 68573 | 49 |
| 07 | 100085 | 100138 | 54 |
| 08 | 102112 | 102203 | 92 |
| 09 | 102281 | 102321 | 41 |
| 10 | 104268 | 104341 | 74 |
| 11 | 106243 | 106299 | 57 |
| a) a) | Hit | Total Info (bits) |
| Hit01 | 85.93 | |
| Hit02 | 144.02 | |
| Hit03 | 90.43 | |
| Hit04 | 81.75 | |
| Hit05 | 82.28 | |
| Hit06 | 83.35 | |
| Hit07 | 90.27 | |
| Hit08 | 165.36 | |
| Hit09 | 73.69 | |
| Hit10 | 125.23 | |
| Hit11 | 95.45 | |
| b) | Ortholog Group | Total Info (bits) |
| Baseline1 | 47.19 | |
| Baseline2 | 70.13 | |
| Baseline3 | 83.94 | |
| Baseline4 | 49.38 | |
| Baseline5 | 77.80 | |
| Baseline6 | 53.79 | |
| Baseline7 | 50.02 | |
| Baseline8 | 71.27 | |
| Baseline9 | 52.13 | |
| Baseline10 | 60.46 | |
| c) c) | Total Info (bits) | |
| t | 3.91 | |
| Std. Deviation | 10.227 | |
| Degrees of Freedom | 19 | |
| p | 0.0009 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2010 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadeque, A.; Barsky, M.; Marass, F.; Kruczkiewicz, P.; Upton, C. JaPaFi: A Novel Program for the Identification of Highly Conserved DNA Sequences. Viruses 2010, 2, 1867-1885. https://doi.org/10.3390/v2091867
Sadeque A, Barsky M, Marass F, Kruczkiewicz P, Upton C. JaPaFi: A Novel Program for the Identification of Highly Conserved DNA Sequences. Viruses. 2010; 2(9):1867-1885. https://doi.org/10.3390/v2091867
Chicago/Turabian StyleSadeque, Aliya, Marina Barsky, Francesco Marass, Peter Kruczkiewicz, and Chris Upton. 2010. "JaPaFi: A Novel Program for the Identification of Highly Conserved DNA Sequences" Viruses 2, no. 9: 1867-1885. https://doi.org/10.3390/v2091867
APA StyleSadeque, A., Barsky, M., Marass, F., Kruczkiewicz, P., & Upton, C. (2010). JaPaFi: A Novel Program for the Identification of Highly Conserved DNA Sequences. Viruses, 2(9), 1867-1885. https://doi.org/10.3390/v2091867