How HIV Takes Advantage of the Cytoskeleton in Entry and Replication


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of the Cytoskeleton in the HIV Life Cycle
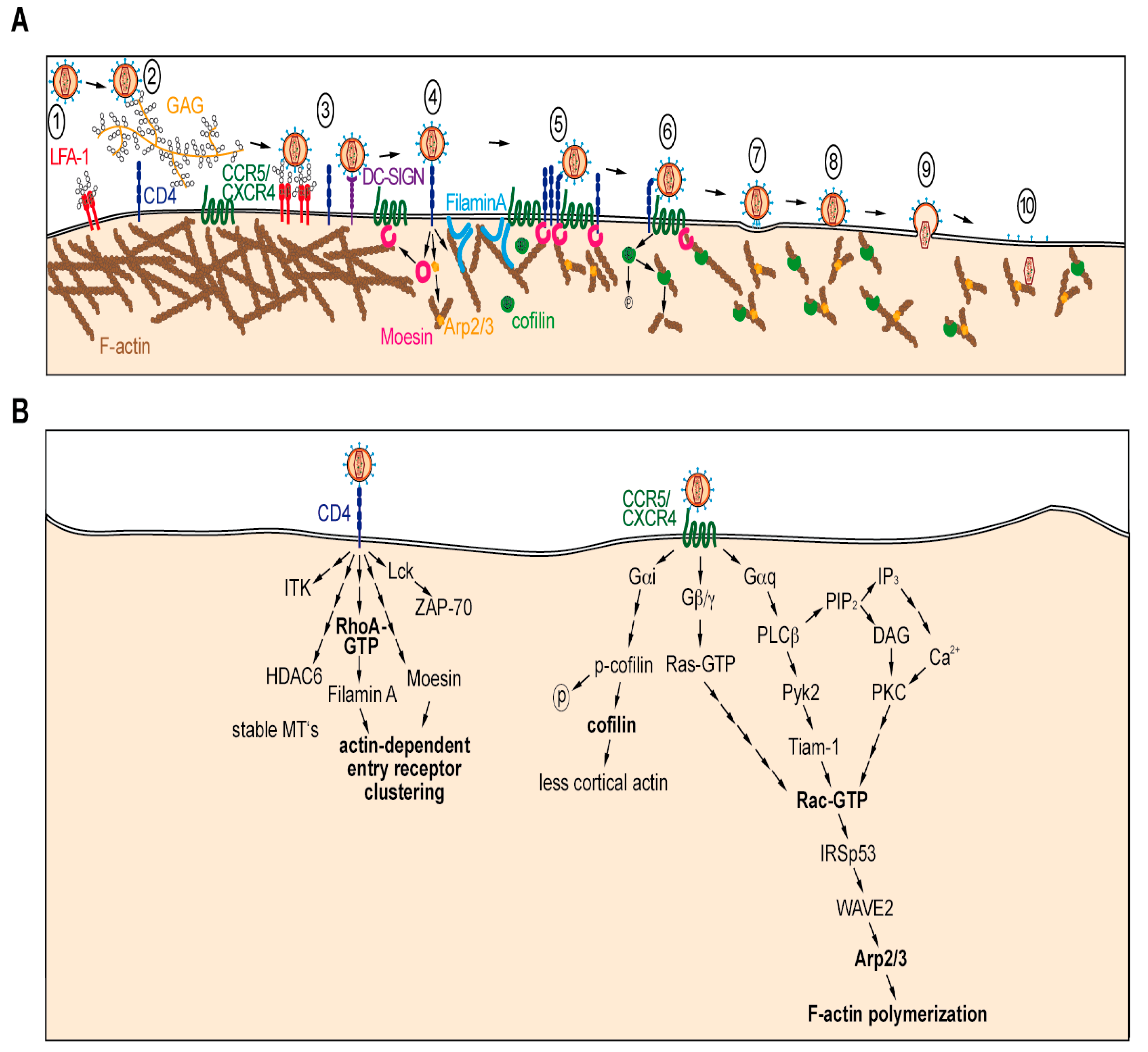
2.1. Entry
2.1.1. Entry Receptor Clustering and Cytoplasmic Delivery of Virion Cores

2.1.2. Sensitization of Uninfected Bystander Cells for Entry
2.1.3. Requirement for Cytoskeletal Interactions during Endocytic Entry?
2.2. Transport to Nucleus
2.3. Transcription and Nuclear Export
2.4. Budding/Assembly/Release
3. HIV-1 Accessory Proteins and Their Influence on the Cytoskeleton:
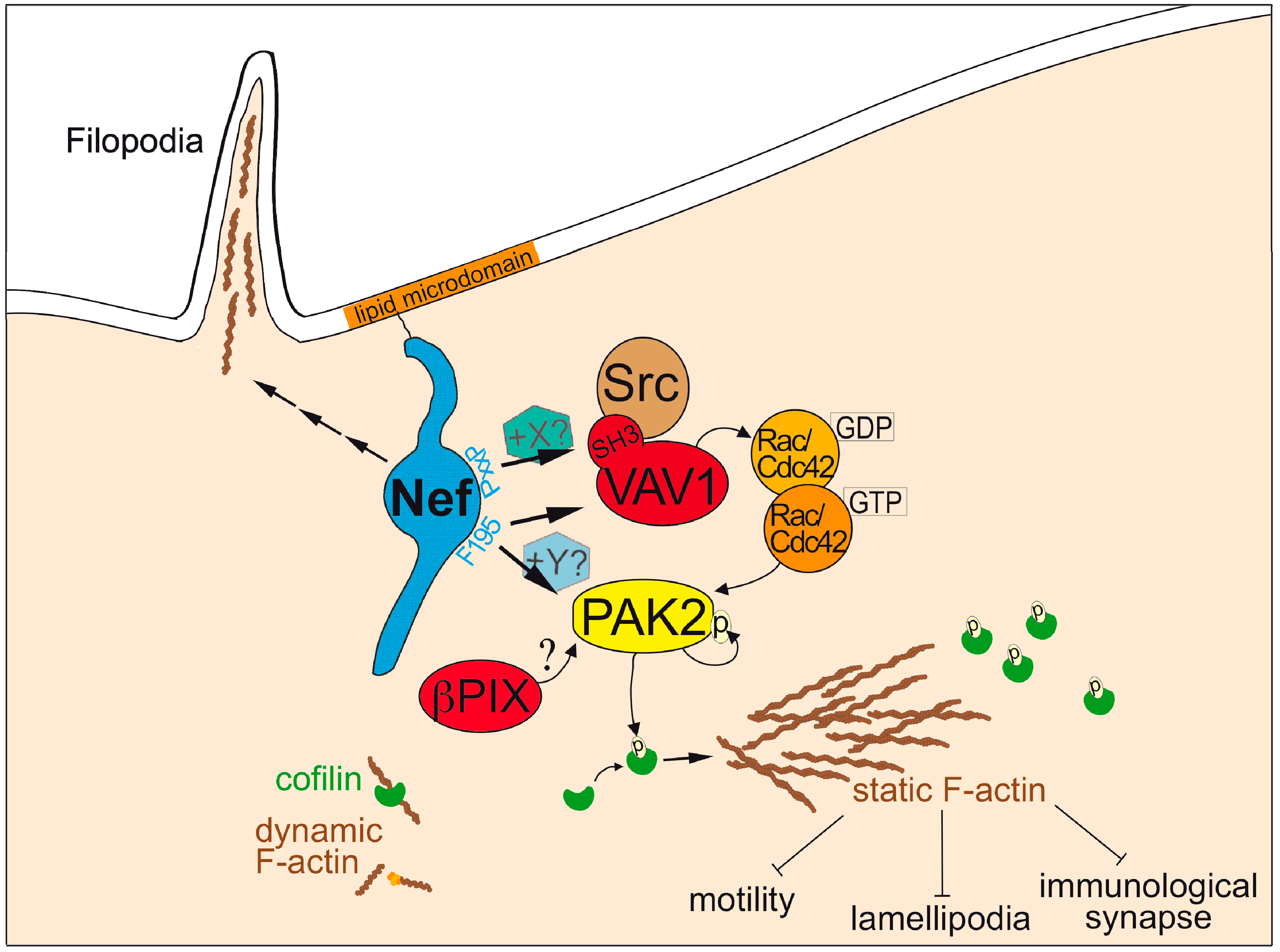
3.1. Neff
3.1.1 Functions of Nef in HIV-1 Infection
3.1.2. Mechanism of Actin Remodeling Inhibition by Nef
3.1.3. Functional Consequences of Altered Actin Dynamics in T Lymphocytes
3.1.4. Nef-Mediated Alteration of F-Actin Organization in Other Cell Types
4. Conclusion
Acknowledgements
References and Notes
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef]
- Chhabra, E.S.; Higgs, H.N. The many faces of actin: Matching assembly factors with cellular structures. Nat. Cell Biol. 2007, 9, 1110–1121. [Google Scholar] [CrossRef]
- Burkhardt, J.K.; Carrizosa, E.; Shaffer, M.H. The actin cytoskeleton in T cell activation. Ann. Rev. Immunol. 2008, 26, 233–259. [Google Scholar] [CrossRef]
- Permanyer, M.; Ballana, E.; Este, J.A. Endocytosis of HIV: Anything goes. Trends Microbiol. 2010, 18, 543–551. [Google Scholar] [CrossRef]
- Doms, R.W.; Trono, D. The plasma membrane as a combat zone in the HIV battlefield. Genes Dev. 2000, 14, 2677–2688. [Google Scholar] [CrossRef]
- Tardif, M.R.; Tremblay, M.J. Regulation of LFA-1 activity through cytoskeleton remodeling and signaling components modulates the efficiency of HIV type-1 entry in activated CD4+ T lymphocytes. J. Immunol. 2005, 175, 926–935. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; Figdor, C.G.; van Kooyk, Y. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Crublet, E.; Andrieu, J.P.; Vives, R.R.; Lortat-Jacob, H. The HIV-1 envelope glycoprotein gp120 features four heparan sulfate binding domains, including the co-receptor binding site. J. Biol. Chem. 2008, 283, 15193–15200. [Google Scholar] [CrossRef]
- Melikyan, G.B. Common principles and intermediates of viral protein-mediated fusion: The HIV-1 paradigm. Retrovirology 2008, 5, 111. [Google Scholar] [CrossRef]
- Iyengar, S.; Hildreth, J.E.; Schwartz, D.H. Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J. Virol. 1998, 72, 5251–5255. [Google Scholar] [CrossRef] [PubMed]
- Steffens, C.M.; Hope, T.J. Mobility of the human immunodeficiency virus (HIV) receptor CD4 and coreceptor CCR5 in living cells: Implications for HIV fusion and entry events. J. Virol. 2004, 78, 9573–9578. [Google Scholar] [CrossRef] [PubMed]
- Barrero-Villar, M.; Cabrero, J.R.; Gordon-Alonso, M.; Barroso-Gonzalez, J.; Alvarez-Losada, S.; Munoz-Fernandez, M.A.; Sanchez-Madrid, F.; Valenzuela-Fernandez, A. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J. Cell Sci. 2009, 122, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Baranda, S.; Gomez-Mouton, C.; Rojas, A.; Martinez-Prats, L.; Mira, E.; Ana Lacalle, R.; Valencia, A.; Dimitrov, D.S.; Viola, A.; Delgado, R.; Martinez, A.C.; Manes, S. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat. Cell Biol. 2007, 9, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell. Biol. 2005, 170, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Harmon, B.; Ratner, L. Induction of the Galpha(q) signaling cascade by the human immunodeficiency virus envelope is required for virus entry. J. Virol. 2008, 82, 9191–9205. [Google Scholar] [CrossRef]
- Yoder, A.; Yu, D.; Dong, L.; Iyer, S.R.; Xu, X.; Kelly, J.; Liu, J.; Wang, W.; Vorster, P.J.; Agulto, L.; Stephany, D.A.; Cooper, J.N.; Marsh, J.W.; Wu, Y. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 2008, 134, 782–792. [Google Scholar] [CrossRef]
- del Real, G.; Jimenez-Baranda, S.; Mira, E.; Lacalle, R.A.; Lucas, P.; Gomez-Mouton, C.; Alegret, M.; Pena, J.M.; Rodriguez-Zapata, M.; Alvarez-Mon, M.; Martinez, A.C.; Manes, S. Statins inhibit HIV-1 infection by down-regulating Rho activity. J. Exp. Med. 2004, 200, 541–547. [Google Scholar] [CrossRef]
- Harmon, B.; Campbell, N.; Ratner, L. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 2010, 6, e1000956. [Google Scholar] [CrossRef]
- Pontow, S.E.; Heyden, N.V.; Wei, S.; Ratner, L. Actin cytoskeletal reorganizations and coreceptor-mediated activation of rac during human immunodeficiency virus-induced cell fusion. J. Virol. 2004, 78, 7138–7147. [Google Scholar] [CrossRef]
- Carter, G.C.; Bernstone, L.; Baskaran, D.; James, W. HIV-1 infects macrophages by exploiting an endocytic route dependent on dynamin, Rac1 and Pak1. Virology 2011, 409, 234–250. [Google Scholar] [CrossRef]
- Komano, J.; Miyauchi, K.; Matsuda, Z.; Yamamoto, N. Inhibiting the Arp2/3 complex limits infection of both intracellular mature vaccinia virus and primate lentiviruses. Mol. Biol. Cell 2004, 15, 5197–5207. [Google Scholar] [CrossRef] [PubMed]
- Vorster, P.J.; Guo, J.; Yoder, A.; Wang, W.; Zheng, Y.; Xu, X.; Yu, D.; Spear, M.; Wu, Y. LIM kinase 1 modulates cortical actin and CXCR4 cycling and is activated by HIV-1 to initiate viral infection. J. Biol. Chem. 2011. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.U.; Saleh, S.; Sallmann, G.; Solomon, A.; Wightman, F.; Evans, V.A.; Boucher, G.; Haddad, E.K.; Sekaly, R.P.; Harman, A.N.; Anderson, J.L.; Jones, K.L.; Mak, J.; Cunningham, A.L.; Jaworowski, A.; Lewin, S.R. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16934–16939. [Google Scholar] [CrossRef]
- Nishita, M.; Aizawa, H.; Mizuno, K. Stromal cell-derived factor 1alpha activates LIM kinase 1 and induces cofilin phosphorylation for T-cell chemotaxis. Mol. Cell Biol. 2002, 22, 774–783. [Google Scholar] [CrossRef]
- Nishita, M.; Tomizawa, C.; Yamamoto, M.; Horita, Y.; Ohashi, K.; Mizuno, K. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J. Cell. Biol. 2005, 171, 349–359. [Google Scholar] [CrossRef]
- Wabnitz, G.H.; Nebl, G.; Klemke, M.; Schroder, A.J.; Samstag, Y. Phosphatidylinositol 3-kinase functions as a Ras effector in the signaling cascade that regulates dephosphorylation of the actin-remodeling protein cofilin after costimulation of untransformed human T lymphocytes. J. Immunol. 2006, 176, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N.; Genovesio, A.; Kim, K.A.; Miko, S.; Perret, E.; Olivo-Marin, J.C.; Shorte, S.; Charneau, P. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 2006, 3, 817–824. [Google Scholar] [CrossRef]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell. Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef]
- Valenzuela-Fernandez, A.; Alvarez, S.; Gordon-Alonso, M.; Barrero, M.; Ursa, A.; Cabrero, J.R.; Fernandez, G.; Naranjo-Suarez, S.; Yanez-Mo, M.; Serrador, J.M.; Munoz-Fernandez, M.A.; Sanchez-Madrid, F. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol. Biol. Cell 2005, 16, 5445–5454. [Google Scholar] [CrossRef]
- Wu, Y.; Yoder, A.; Yu, D.; Wang, W.; Liu, J.; Barrett, T.; Wheeler, D.; Schlauch, K. Cofilin activation in peripheral CD4 T cells of HIV-1 infected patients: A pilot study. Retrovirology 2008, 5, 95. [Google Scholar] [CrossRef]
- Fackler, O.T.; Peterlin, B.M. Endocytic entry of HIV-1. Curr. Biol. 2000, 10, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Krausslich, H.G. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 2005, 79, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.; Grigorov, B.; Senserrich, J.; Clotet, B.; Darlix, J.L.; Muriaux, D.; Este, J.A. A clathrin-dynamin-dependent endocytic pathway for the uptake of HIV-1 by direct T cell-T cell transmission. Antivir. Res. 2008, 80, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Clotet-Codina, I.; Bosch, B.; Senserrich, J.; Fernandez-Figueras, M.T.; Pena, R.; Ballana, E.; Bofill, M.; Clotet, B.; Este, J.A. HIV endocytosis after dendritic cell to T cell viral transfer leads to productive virus infection. Antivir. Res. 2009, 83, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Marechal, V.; Prevost, M.C.; Petit, C.; Perret, E.; Heard, J.M.; Schwartz, O. Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J. Virol. 2001, 75, 11166–11177. [Google Scholar] [CrossRef]
- Kaksonen, M.; Toret, C.P.; Drubin, D.G. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat. Rev. Mol. Cell. Biol. 2006, 7, 404–414. [Google Scholar] [CrossRef]
- Robertson, A.S.; Smythe, E.; Ayscough, K.R. Functions of actin in endocytosis. Cell. Mol. Life Sci. 2009, 66, 2049–2065. [Google Scholar] [CrossRef]
- Conibear, E. Converging views of endocytosis in yeast and mammals. Curr. Opin. Cell Biol. 2010, 22, 513–518. [Google Scholar] [CrossRef]
- Haller, C.; Fackler, O.T. HIV-1 at the immunological and T-lymphocytic virological synapse. Biol. Chem. 2008, 389, 1253–1560. [Google Scholar] [CrossRef]
- Jin, J.; Sherer, N.M.; Heidecker, G.; Derse, D.; Mothes, W. Assembly of the murine leukemia virus is directed towards sites of cell-cell contact. PLoS Biol. 2009, 7, e1000163. [Google Scholar] [CrossRef]
- Feldmann, J.; Schwartz, O. HIV-1 Virological Synapse: Live Imaging of Transmission. Viruses 2010, 2, 1666–1680. [Google Scholar] [CrossRef]
- Sattentau, Q.J. Retroviruses and the Third Synapse. Viruses 2010, 2, 1008–1010. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N. Revisiting HIV-1 uncoating. Retrovirology 2010, 7, 96. [Google Scholar] [CrossRef]
- Arhel, N.J.; Souquere-Besse, S.; Munier, S.; Souque, P.; Guadagnini, S.; Rutherford, S.; Prevost, M.C.; Allen, T.D.; Charneau, P. HIV-1 DNA Flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 2007, 26, 3025–3037. [Google Scholar] [CrossRef] [PubMed]
- Dismuke, D.J.; Aiken, C. Evidence for a functional link between uncoating of the human immunodeficiency virus type 1 core and nuclear import of the viral preintegration complex. J. Virol. 2006, 80, 3712–3720. [Google Scholar] [CrossRef] [PubMed]
- Dvorin, J.D.; Malim, M.H. Intracellular trafficking of HIV-1 cores: Journey to the center of the cell. Curr. Top. Microbiol. Immunol. 2003, 281, 179–208. [Google Scholar] [PubMed]
- Bukrinskaya, A.; Brichacek, B.; Mann, A.; Stevenson, M. Establishment of a functional human immunodeficiency virus type 1 (HIV-1) reverse transcription complex involves the cytoskeleton. J. Exp. Med. 1998, 188, 2113–2125. [Google Scholar] [CrossRef]
- Wiskerchen, M.; Muesing, M.A. Human immunodeficiency virus type 1 integrase: Effects of mutations on viral ability to integrate, direct viral gene expression from unintegrated viral DNA templates, and sustain viral propagation in primary cells. J. Virol. 1995, 69, 376–386. [Google Scholar] [CrossRef]
- Peterlin, B.M. Transcription elongation takes central stage: The P-TEFb connection. Cell Cycle 2010, 9, 2933–2934. [Google Scholar] [CrossRef]
- Brandt, D.T.; Xu, J.; Steinbeisser, H.; Grosse, R. Regulation of myocardin-related transcriptional coactivators through cofactor interactions in differentiation and cancer. Cell Cycle 2009, 8, 2523–2527. [Google Scholar] [CrossRef] [PubMed]
- Vartiainen, M.K. Nuclear actin dynamics--from form to function. FEBS Lett. 2008, 582, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Han, M.; Bernier, M.; Wen, J.K. Nuclear actin and actin-binding proteins in the regulation of transcription and gene expression. FEBS J. 2009, 276, 2669–2685. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.F.; Gu, Y.; Xu, Y.C.; Mitola, S.; Bussolino, F.; Terada, L.S. Human immunodeficiency virus type 1 Tat regulates endothelial cell actin cytoskeletal dynamics through PAK1 activation and oxidant production. J. Virol. 2004, 78, 779–789. [Google Scholar] [CrossRef]
- Lopez-Huertas, M.R.; Callejas, S.; Abia, D.; Mateos, E.; Dopazo, A.; Alcami, J.; Coiras, M. Modifications in host cell cytoskeleton structure and function mediated by intracellular HIV-1 Tat protein are greatly dependent on the second coding exon. Nucl. Acid. Res. 2010, 38, 3287–3307. [Google Scholar] [CrossRef]
- Benkirane, M.; Chun, R.F.; Xiao, H.; Ogryzko, V.V.; Howard, B.H.; Nakatani, Y.; Jeang, K.T. Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 1998, 273, 24898–24905. [Google Scholar] [CrossRef]
- Mahmoudi, T.; Parra, M.; Vries, R.G.; Kauder, S.E.; Verrijzer, C.P.; Ott, M.; Verdin, E. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 2006, 281, 19960–19968. [Google Scholar] [CrossRef]
- Farrants, A.K. Chromatin remodelling and actin organisation. FEBS Lett. 2008, 582, 2041–2050. [Google Scholar] [CrossRef]
- Kimura, T.; Hashimoto, I.; Yamamoto, A.; Nishikawa, M.; Fujisawa, J.I. Rev-dependent association of the intron-containing HIV-1 gag mRNA with the nuclear actin bundles and the inhibition of its nucleocytoplasmic transport by latrunculin-B. Genes Cells 2000, 5, 289–307. [Google Scholar] [CrossRef]
- Audoly, G.; Popoff, M.R.; Gluschankof, P. Involvement of a small GTP binding protein in HIV-1 release. Retrovirology 2005, 2, 48. [Google Scholar] [CrossRef]
- Sasaki, H.; Nakamura, M.; Ohno, T.; Matsuda, Y.; Yuda, Y.; Nonomura, Y. Myosin-actin interaction plays an important role in human immunodeficiency virus type 1 release from host cells. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2026–2030. [Google Scholar] [CrossRef]
- Müller, B.; Kräusslich, H.-G. Actin-disrupting drugs do not affect the localization of HIV-1 Gag or kinetics of budding. Department of Infectious Diseases, Virology, Heidelberg, Germany, 2011, Unpublished observations.
- Chukkapalli, V.; Hogue, I.B.; Boyko, V.; Hu, W.S.; Ono, A. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J. Virol. 2008, 82, 2405–2417. [Google Scholar] [CrossRef]
- Tang, C.; Loeliger, E.; Luncsford, P.; Kinde, I.; Beckett, D.; Summers, M.F. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 517–522. [Google Scholar] [CrossRef]
- Liu, B.; Dai, R.; Tian, C.J.; Dawson, L.; Gorelick, R.; Yu, X.F. Interaction of the human immunodeficiency virus type 1 nucleocapsid with actin. J. Virol. 1999, 73, 2901–2908. [Google Scholar] [CrossRef]
- Rey, O.; Canon, J.; Krogstad, P. HIV-1 Gag protein associates with F-actin present in microfilaments. Virology 1996, 220, 530–534. [Google Scholar] [CrossRef]
- Shoeman, R.L.; Kesselmier, C.; Mothes, E.; Honer, B.; Traub, P. Non-viral cellular substrates for human immunodeficiency virus type 1 protease. FEBS Lett. 1991, 278, 199–203. [Google Scholar] [CrossRef]
- Martinez, N.W.; Xue, X.; Berro, R.G.; Kreitzer, G.; Resh, M.D. Kinesin KIF4 regulates intracellular trafficking and stability of the human immunodeficiency virus type 1 Gag polyprotein. J. Virol. 2008, 82, 9937–9950. [Google Scholar] [CrossRef]
- Swanson, C.M.; Malim, M.H. Retrovirus RNA trafficking: From chromatin to invasive genomes. Traffic 2006, 7, 1440–1450. [Google Scholar] [CrossRef]
- Gladnikoff, M.; Shimoni, E.; Gov, N.S.; Rousso, I. Retroviral assembly and budding occur through an actin-driven mechanism. Biophys. J. 2009, 97, 2419–2428. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Johnson, D.G.; Kane, B.P.; Sowder, R.C., 2nd; Kim, Y.D.; Fisher, R.J.; Zhou, X.Z.; Lu, K.P. Henderson, L.E. Actin-binding cellular proteins inside human immunodeficiency virus type 1. Virology 2000, 266, 42–51. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Kane, B.P.; Busch, L.K.; Johnson, D.G.; Sowder, R.C., 2nd; Chertova, E.N.; Arthur, L.O.; Henderson, L.E. Cytoskeletal proteins inside human immunodeficiency virus type 1 virions. J. Virol. 1996, 70, 7734–7743. [Google Scholar] [CrossRef]
- Bieniasz, P.D. The cell biology of HIV-1 virion genesis. Cell Host Microbe 2009, 5, 550–558. [Google Scholar] [CrossRef]
- Carlton, J.G.; Agromayor, M.; Martin-Serrano, J. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10541–10546. [Google Scholar] [CrossRef]
- Sevrioukov, E.A.; Moghrabi, N.; Kuhn, M.; Kramer, H. A mutation in dVps28 reveals a link between a subunit of the endosomal sorting complex required for transport-I complex and the actin cytoskeleton in Drosophila. Mol. Biol. Cell. 2005, 16, 2301–2312. [Google Scholar] [CrossRef]
- Karczewski, M.K.; Strebel, K. Cytoskeleton association and virion incorporation of the human immunodeficiency virus type 1 Vif protein. J. Virol. 1996, 70, 494–507. [Google Scholar] [CrossRef]
- Goila-Gaur, R.; Strebel, K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 2008, 5, 51. [Google Scholar] [CrossRef]
- Kestler, H.W., 3rd; Ringler, D.J.; Mori, K.; Panicali, D.L.; Sehgal, P.K.; Daniel, M.D.; Desrosiers, R.C. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 1991, 65, 651–662. [Google Scholar] [CrossRef]
- Geyer, M.; Fackler, O.T.; Peterlin, B.M. Structure-function relationships in HIV-1 Nef. EMBO Rep. 2001, 2, 580–585. [Google Scholar] [CrossRef]
- Laguette, N.; Bregnard, C.; Benichou, S.; Basmaciogullari, S. Human immunodeficiency virus (HIV) type-1, HIV-2 and simian immunodeficiency virus Nef proteins. Mol. Aspects Med. 2010, 31, 418–433. [Google Scholar] [CrossRef]
- Arhel, N.J.; Kirchhoff, F. Implications of Nef: Host cell interactions in viral persistence and progression to AIDS. Curr. Top. Microbiol. Immunol. 2009, 339, 147–175. [Google Scholar]
- Fackler, O.T.; Luo, W.; Geyer, M.; Alberts, A.S.; Peterlin, B.M. Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol. Cell 1999, 3, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Reichman-Fried, M.; Abraham, L.; Pan, X.; Giese, S.I.; Hannemann, S.; Goulimari, P.; Raz, E.; Grosse, R.; Fackler, O.T. HIV-1 Nef interferes with host cell motility by deregulation of Cofilin. Cell Host Microbe 2009, 6, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Rauch, S.; Fackler, O.T. HIV-1 Nef employs two distinct mechanisms to modulate Lck subcellular localization and TCR induced actin remodeling. PLoS ONE 2007, 2, e1212. [Google Scholar] [CrossRef]
- Haller, C.; Rauch, S.; Michel, N.; Hannemann, S.; Lehmann, M.J.; Keppler, O.T.; Fackler, O.T. The HIV-1 pathogenicity factor Nef interferes with maturation of stimulatory T-lymphocyte contacts by modulation of N-Wasp activity. J. Biol. Chem. 2006, 281, 19618–19630. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Rajan, D.; Specht, A.; Ritter, C.; Pulkkinen, K.; Saksela, K.; Kirchhoff, F. Association of Nef with p21-activated kinase 2 is dispensable for efficient human immunodeficiency virus type 1 replication and cytopathicity in ex vivo-infected human lymphoid tissue. J. Virol. 2007, 81, 13005–13014. [Google Scholar] [CrossRef]
- Stolp, B.; Abraham, L.; Rudolph, J.M.; Fackler, O.T. Lentiviral Nef proteins utilize PAK2-mediated deregulation of cofilin as a general strategy to interfere with actin remodeling. J. Virol. 2010, 84, 3935–3948. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Pulkkinen, K.; Saksela, K.; Fackler, O.T. Human immunodeficiency virus type 1 Nef recruits the guanine exchange factor Vav1 via an unexpected interface into plasma membrane microdomains for association with p21-activated kinase 2 activity. J. Virol. 2008, 82, 2918–2929. [Google Scholar] [CrossRef]
- Krautkramer, E.; Giese, S.I.; Gasteier, J.E.; Muranyi, W.; Fackler, O.T. Human immunodeficiency virus type 1 Nef activates p21-activated kinase via recruitment into lipid rafts. J. Virol. 2004, 78, 4085–4097. [Google Scholar] [CrossRef]
- Pulkkinen, K.; Renkema, G.H.; Kirchhoff, F.; Saksela, K. Nef associates with p21-activated kinase 2 in a p21-GTPase-dependent dynamic activation complex within lipid rafts. J. Virol. 2004, 78, 12773–12780. [Google Scholar] [CrossRef]
- Agopian, K.; Wei, B.L.; Garcia, J.V.; Gabuzda, D. A hydrophobic binding surface on the human immunodeficiency virus type 1 Nef core is critical for association with p21-activated kinase 2. J. Virol. 2006, 80, 3050–3061. [Google Scholar] [CrossRef]
- Manninen, A.; Hiipakka, M.; Vihinen, M.; Lu, W.; Mayer, B.J.; Saksela, K. SH3-Domain binding function of HIV-1 Nef is required for association with a PAK-related kinase. Virology 1998, 250, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Manninen, A.; Saksela, K. Human immunodeficiency virus type 1 Nef selectively associates with a catalytically active subpopulation of p21-activated kinase 2 (PAK2) independently of PAK2 binding to Nck or beta-PIX. J. Virol. 2001, 75, 2154–2160. [Google Scholar] [CrossRef]
- Rudolph, J.M.; Eickel, N.; Haller, C.; Schindler, M.; Fackler, O.T. Inhibition of T-cell receptor-induced actin remodeling and relocalization of Lck are evolutionarily conserved activities of lentiviral Nef proteins. J. Virol. 2009, 83, 11528–11539. [Google Scholar] [CrossRef]
- Brown, A.; Wang, X.; Sawai, E.; Cheng-Mayer, C. Activation of the PAK-related kinase by human immunodeficiency virus type 1 Nef in primary human peripheral blood lymphocytes and macrophages leads to phosphorylation of a PIX-p95 complex. J. Virol. 1999, 73, 9899–9907. [Google Scholar] [CrossRef]
- Vincent, P.; Priceputu, E.; Kay, D.; Saksela, K.; Jolicoeur, P.; Hanna, Z. Activation of p21-activated kinase 2 and its association with Nef are conserved in murine cells but are not sufficient to induce an AIDS-like disease in CD4C/HIV transgenic mice. J. Biol. Chem. 2006, 281, 6940–6954. [Google Scholar] [CrossRef]
- Nobile, C.; Rudnicka, D.; Hasan, M.; Aulner, N.; Porrot, F.; Machu, C.; Renaud, O.; Prevost, M.C.; Hivroz, C.; Schwartz, O.; Sol-Foulon, N. HIV-1 Nef inhibits ruffles, induces filopodia and modulates migration of infected lymphocytes. J. Virol. 2009, 84, 2282–2293. [Google Scholar] [CrossRef]
- Haller, C.; Tibroni, N.; Rudolph, J.M.; Grosse, R.; Fackler, O.T. Nef does not inhibit F-actin remodelling and HIV-1 cell-cell transmission at the T lymphocyte virological synapse. Eur. J. Cell Biol. 2010. [Google Scholar] [CrossRef]
- Friedl, P.; den Boer, A.T.; Gunzer, M. Tuning immune responses: Diversity and adaptation of the immunological synapse. Nat. Rev. Immunol. 2005, 5, 532–545. [Google Scholar] [CrossRef]
- Arhel, N.; Lehmann, M.; Clauss, K.; Nienhaus, G.U.; Piguet, V.; Kirchhoff, F. The inability to disrupt the immunological synapse between infected human T cells and APCs distinguishes HIV-1 from most other primate lentiviruses. J. Clin. Invest. 2009, 119, 2965–2975. [Google Scholar] [CrossRef]
- Thoulouze, M.I.; Sol-Foulon, N.; Blanchet, F.; Dautry-Varsat, A.; Schwartz, O.; Alcover, A. Human immunodeficiency virus type-1 infection impairs the formation of the immunological synapse. Immunity 2006, 24, 547–561. [Google Scholar] [CrossRef]
- Schindler, M.; Munch, J.; Kutsch, O.; Li, H.; Santiago, M.L.; Bibollet-Ruche, F.; Muller-Trutwin, M.C.; Novembre, F.J.; Peeters, M.; Courgnaud, V.; Bailes, E.; Roques, P.; Sodora, D.L.; Silvestri, G.; Sharp, P.M.; Hahn, B.H.; Kirchhoff, F. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 2006, 125, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Alcover, A.; Schwartz, O. Modulation of the immunological synapse: A key to HIV-1 pathogenesis? Nat. Rev. Immunol. 2007, 7, 310–317. [Google Scholar] [CrossRef]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Choe, E.Y.; Schoenberger, E.S.; Groopman, J.E.; Park, I.W. HIV Nef inhibits T cell migration. J. Biol. Chem. 2002, 277, 46079–46084. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.V.; Nombela-Arrieta, C. Chemokine control of lymphocyte trafficking: A general overview. Immunology 2005, 116, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Gray, E.E.; Cyster, J.G. The microanatomy of B cell activation. Curr. Opin. Immunol. 2009, 21, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, C.; Tadakuma, K.; Otani, I.; Moritoyo, T.; Akari, H.; Ono, F.; Yoshikawa, Y.; Sata, T.; Izumo, S.; Mori, K. nef gene is required for robust productive infection by simian immunodeficiency virus of T-cell-rich paracortex in lymph nodes. J. Virol. 2003, 77, 4169–4180. [Google Scholar] [CrossRef]
- Moir, S.; Fauci, A.S. B cells in HIV infection and disease. Nat. Rev. Immunol. 2009, 9, 235–245. [Google Scholar] [CrossRef]
- Schacker, T. The role of secondary lymphatic tissue in immune deficiency of HIV infection. AIDS 2008, 22, S13–S18. [Google Scholar] [CrossRef]
- Sourisseau, M.; Sol-Foulon, N.; Porrot, F.; Blanchet, F.; Schwartz, O. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 2007, 81, 1000–1012. [Google Scholar] [CrossRef]
- Sato, H.; Orenstein, J.; Dimitrov, D.; Martin, M. Cell-to-cell spread of HIV-1 occurs within minutes and may not involve the participation of virus particles. Virology 1992, 186, 712–724. [Google Scholar] [CrossRef]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Kohler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; Onfelt, B.; Sattentau, Q.; Davis, D.M. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Santini, P.A.; Sullivan, J.S.; He, B.; Shan, M.; Ball, S.C.; Dyer, W.B.; Ketas, T.J.; Chadburn, A.; Cohen-Gould, L.; Knowles, D.M.; Chiu, A.; Sanders, R.W.; Chen, K.; Cerutti, A. HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long-range intercellular conduits. Nat. Immunol. 2009, 10, 1008–1017. [Google Scholar] [CrossRef]
- Qiao, X.; He, B.; Chiu, A.; Knowles, D.M.; Chadburn, A.; Cerutti, A. Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in bystander B cells. Nat. Immunol. 2006, 7, 302–310. [Google Scholar] [CrossRef]
- Swingler, S.; Zhou, J.; Swingler, C.; Dauphin, A.; Greenough, T.; Jolicoeur, P.; Stevenson, M. Evidence for a pathogenic determinant in HIV-1 Nef involved in B cell dysfunction in HIV/AIDS. Cell Host Microbe 2008, 4, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.C.; He, J.C.; Wang, Z.H.; Feng, X.; Fukumi-Tominaga, T.; Chen, N.; Xu, J.; Iyengar, R.; Klotman, P.E. HIV-1 Nef disrupts the podocyte actin cytoskeleton by interacting with diaphanous interacting protein. J. Biol. Chem. 2008, 283, 8173–8182. [Google Scholar] [CrossRef]
- Quaranta, M.G.; Mattioli, B.; Spadaro, F.; Straface, E.; Giordani, L.; Ramoni, C.; Malorni, W.; Viora, M. HIV-1 Nef triggers Vav-mediated signaling pathway leading to functional and morphological differentiation of dendritic cells. FASEB J. 2003, 17, 2025–2036. [Google Scholar] [CrossRef]

© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolp, B.; Fackler, O.T. How HIV Takes Advantage of the Cytoskeleton in Entry and Replication. Viruses 2011, 3, 293-311. https://doi.org/10.3390/v3040293
Stolp B, Fackler OT. How HIV Takes Advantage of the Cytoskeleton in Entry and Replication. Viruses. 2011; 3(4):293-311. https://doi.org/10.3390/v3040293
Chicago/Turabian StyleStolp, Bettina, and Oliver T. Fackler. 2011. "How HIV Takes Advantage of the Cytoskeleton in Entry and Replication" Viruses 3, no. 4: 293-311. https://doi.org/10.3390/v3040293
APA StyleStolp, B., & Fackler, O. T. (2011). How HIV Takes Advantage of the Cytoskeleton in Entry and Replication. Viruses, 3(4), 293-311. https://doi.org/10.3390/v3040293