Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus
by
Sharmi W. Thor
1, 
Deborah A. Hilt
1,
Jessica C. Kissinger
2,
Andrew H. Paterson
3 and
Mark W. Jackwood
1,* 1
Department of Population Health, Poultry Diagnostic and Research Center, College of Veterinary Medicine, University of Georgia, Athens, GA 30602, USA
2
Department of Genetics, Center for Tropical and Emerging Global Diseases, University of Georgia, Athens, GA 30602, USA
3
Plant Genome Mapping Laboratory, Departments of Crop and Soil Sciences, Plant Biology and Genetics, University of Georgia, Athens, GA 30602, USA
*
Author to whom correspondence should be addressed.
Viruses 2011, 3(9), 1777-1799; https://doi.org/10.3390/v3091777
Submission received: 8 August 2011
/
Revised: 24 August 2011
/
Accepted: 5 September 2011
/
Published: 23 September 2011
(This article belongs to the Special Issue Recombination in Viruses)
Abstract
:Recombination in the family Coronaviridae has been well documented and is thought to be a contributing factor in the emergence and evolution of different coronaviral genotypes as well as different species of coronavirus. However, there are limited data available on the frequency and extent of recombination in coronaviruses in nature and particularly for the avian gamma-coronaviruses where only recently the emergence of a turkey coronavirus has been attributed solely to recombination. In this study, the full-length genomes of eight avian gamma-coronavirus infectious bronchitis virus (IBV) isolates were sequenced and along with other full-length IBV genomes available from GenBank were analyzed for recombination. Evidence of recombination was found in every sequence analyzed and was distributed throughout the entire genome. Areas that have the highest occurrence of recombination are located in regions of the genome that code for nonstructural proteins 2, 3 and 16, and the structural spike glycoprotein. The extent of the recombination observed, suggests that this may be one of the principal mechanisms for generating genetic and antigenic diversity within IBV. These data indicate that reticulate evolutionary change due to recombination in IBV, likely plays a major role in the origin and adaptation of the virus leading to new genetic types and strains of the virus.
1. Introduction
Avian infectious bronchitis virus (IBV) is a gamma-coronavirus in the family Coronaviridae, the order Nidovirales, and the genus Coronavirus that causes a highly contagious upper-respiratory disease of domestic chickens. In layer type birds it can cause a drop in egg production and some strains are nephropathogenic. Infectious bronchitis remains one of the most widely reported respiratory diseases of chickens worldwide despite the routine usage of attenuated live vaccines to control the disease. Control of IBV is difficult because there is little to no cross-protection between the numerous different serotypes of the virus.
Infectious bronchitis virus is an enveloped, single-stranded, positive-sense RNA virus with a genome length of approximately 27 kb. The 3’ end of the genome encodes four structural proteins; spike (S), envelope (E), membrane (M) and nucleocapsid (N) as well as several non-structural proteins [1]. The S glycoprotein of IBV forms projections on the surface of the virion. Spike is post-translationally cleaved into S1 and S2 subunits with the S1 subunit forming the outermost portion and S2 forming a stalk-like structure that is embedded in the viral membrane. The S1 subunit contains hypervariable regions that play a role in attachment to host cell receptors, and it contains conformationally-dependent virus-neutralizing and serotype-specific epitopes [2,3]. Spike is also involved in membrane fusion and viral entry into the host cell. The E and M proteins are integral membrane proteins involved in assembly of the virus. The N protein is closely associated with the viral genome and plays a role in replication. The 5’ two-thirds of the genome, approximately 21 kb, encodes two polyproteins 1a and 1ab. A minus one frame-shift mechanism is used to translate the 1ab polyprotein. The polyproteins are post-translationally cleaved into 15 non-structural proteins (nsps), nsp 2–16 (IBV does not have an nsp1) that make up the replication complex. Key nsps encoded, include a papain-like protease 2 (PLP2) within nsp 3, a main protease (Mpro) within nsp 5, and the RNA-dependent RNA-polymerase (RdRp) within nsps 11 and 12.
Genetic diversity in coronaviruses is due to adaptive evolution driven by high mutation rates and genetic recombination [4]. High mutation rates are attributed to minimal proof reading capabilities associated with the RdRp. Recombination is thought to be due to a unique template switching “copy-choice” mechanism during RNA replication [5]. Evidence of recombination among strains of IBV has been observed both experimentally and in the field [6,7,8,9,10,11]. The emergence of several alpha- and beta-coronaviruses has been attributed to recombination [12,13] but only recently was recombination shown to be the mechanism behind the emergence of a novel gamma-coronavirus, turkey coronavirus (TCoV) [14]. Although “hot spots” of recombination in the genome of IBV have been reported [9,15], a thorough study of recombination using multiple different strains across the entire genome has not been conducted.
In this study we sequenced and analyzed the entire genome of eight IBV strains that represent different serotypes that have not been previously sequenced, and we compared these sequences with other gamma-coronavirus full-length genome sequences available in GenBank for evidence of recombination [16]. Different serotypes of field viruses and vaccine type viruses were selected to provide a wide variety of sequences potentially capable of contributing gene fragments to recombinants.
2. Results and Discussion
2.1. Sequence Analysis
The full-length genomes of eight isolates of IBV were sequenced at 5× to 10× coverage, and the consensus sequences were assembled. The genome size (see the end of the 3’UTR in Table 1), organization of the genome, and the location and size of the open reading frames (ORFs) are listed in Table 1 for each of the viruses. The gene order is the same for all the viruses examined; 5’UTR-1a/ab-spike-3a-3b-Envelope-Membrane-4b-4c-5a-5b-Nucleocapsid-3’UTR. In addition, the genomes for CAV/CAV56b/91, DE/DE072/92, FL/FL18288/71, Mass/H120, Iowa/Iowa97/56 and JMK/JMK/64 contain ORF 6b between nucleocapsid and the 3’UTR.
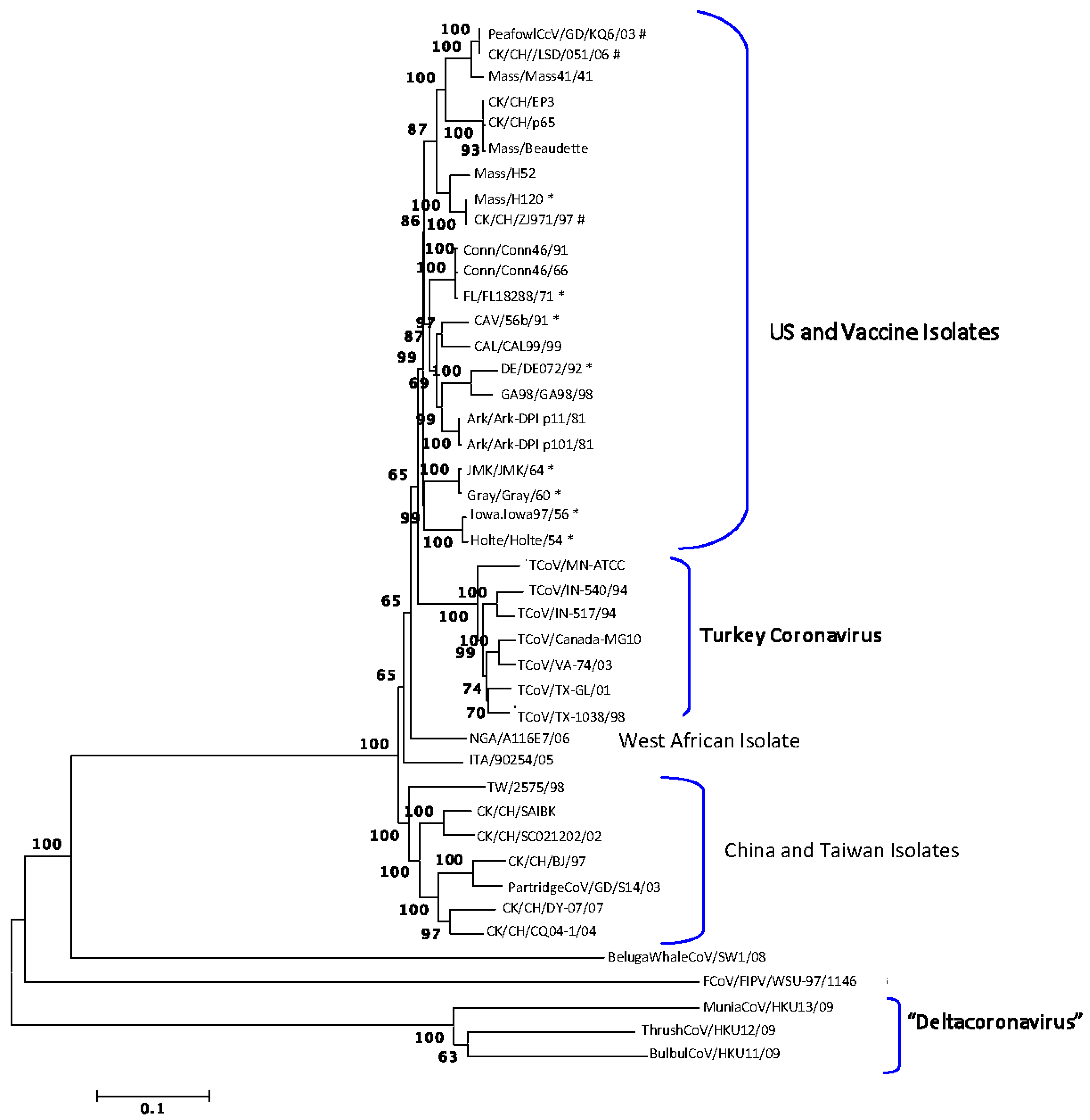
The full-length genomes were aligned and phylogenetic trees were constructed using the Neighbor-joining, Minimum Evolution, Maximum Parsimony and UPGMA programs in MEGA4 [17]. The trees all had similar topology and bootstrap support, and a representative tree is shown in Figure 1. The feline coronavirus FCoV/FIPV/WSU-79-1146 and the beluga whale virus BelugaWhaleCoV/SW1/08 were included as out-groups. The wild bird viruses isolated from a munia (MuniaCoV/HKUY13/09), thrush (ThrushCoV/HKU12/09) and bulbul (BulBulCoV/HKU11/09) formed a unique clade, which is not surprising as this group might represent a new coronavirus genus provisionally designated Deltacoronavirus [18]. The remaining viruses separated into clades consisting of IBV isolates from the US and vaccine viruses, TCoV isolates, an IBV isolate from West Africa and IBV isolates from China and Taiwan.
Vaccines for IBV used in commercial poultry include the serotypes Mass, Conn, DE and Ark. The PeafowlCcV/GD/KQ6/03, CK/CH/LSD/051/06 and CK/CH/ZJ971/97 strains from China grouped with Mass type viruses indicating that they are closely related, which is not surprising since Mass type vaccines are used in China. The overall percent similarities between the various strains are listed in Supplemental Table 1. All IBV genomes examined are greater than 80% similar at the nucleotide level.
2.2. Recombination Analysis
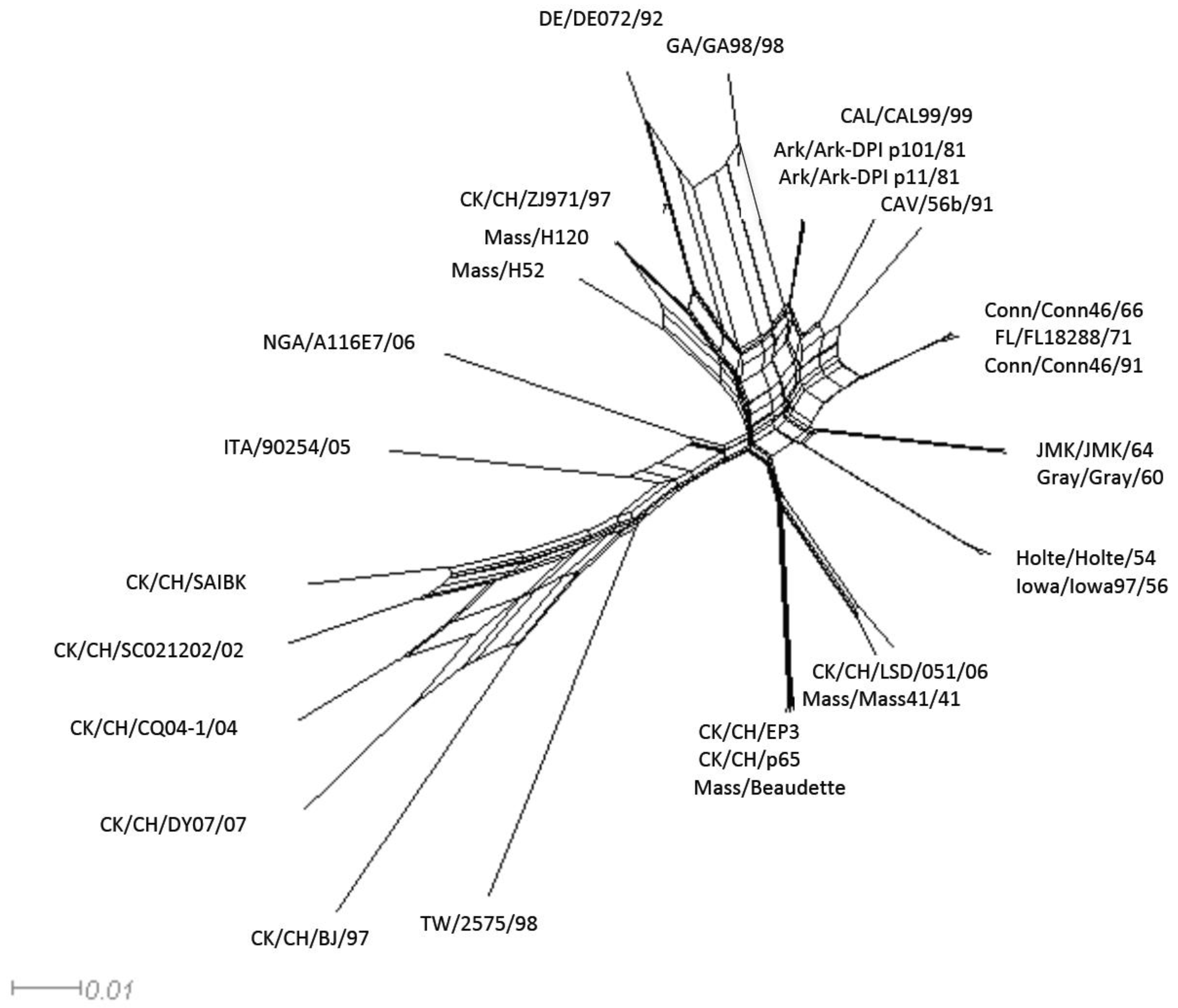
Recombination among coronaviruses reduces mutational load, creates genetic variation, and can result in the emergence of new strains [19]. However, evolutionary history is traditionally represented using a strictly bifurcating phylogenetic tree, which implies that once two lineages are created they subsequently never interact with each other. When evolutionary events such as reassortment, horizontal gene transfer or recombination occur, reticulations among the phylogenetic tree branches can result. Using the Neighbor-net analysis we observed evidence of networked relationships (represented by boxes, in Figure 2) among the analyzed sequences. Since the boxes only imply the possibility of recombination, we conducted a pairwise homoplasy index (PHI) test, which showed a significant difference in the compatibility between closely linked sites (p < 0.0001) supporting the occurrences of recombination among the viruses [20].
The Recombination Detection Program 4 (RDP4) [21,22] was used to identify recombination breakpoint positions in full-length IBV genome sequences and the data for 50 of a total 135 unique transferred fragments with statistical support of p ≤ 1 × 10−12 are listed in Table 2. The full-length genomes available in the database for CK/CH/EP3, CK/CH/p65, and Mass/Beaudette were excluded from the analysis because they are viruses not found in the field. The recombination programs can be used to detect recombination without reference sequences, and our analysis was conducted without regard to date of isolation because that information was not available for some of the viruses. Although the programs attempt to identify major and minor parent sequences contributing to each recombinant, the data reported herein only represents sequences in other viruses that are most closely related to the sequence surrounding the transferred fragment (major sequence) and the sequence closely related to the transferred fragment (minor sequences) and doesn’t imply origin or source of the transferred fragment. In many cases, the transferred fragment has undergone mutations making it difficult to identify all the endpoints for the major and minor sequences. In addition, some of the transferred fragments overlap suggesting that recombinations have occurred between recombinant viruses.
Twenty-five IBV strains were examined and the viruses with the most transferred fragments in Table 2 are CAV/56b/91 and Mass/H52 both with 8 fragments, and CK/CH/LSD/051/06 and GA98/0470/98 both with 7 fragments. The strains with the fewest transferred fragments are Iowa/Iowa97/56 and TW/2575/98 with only 2 transferred fragments and the CK/CH/BJ/97, Holte/Holte/54, and NGA/A116E7/06 strains with only 1 transferred fragment. The Ark/Ark-DPI-p11/81 and Ark/Ark-DPI-p101/81 strains are the same virus that was passaged 11 and 101 times in embryonated eggs, respectively. Both viruses share identical transferred fragments indicating that they have identical recombination history. In addition, Conn/Conn46/66 and Conn/Conn46/91 share the same recombination history (4 identical transferred fragments). The Conn/Conn46/66 field virus was used to produce an attenuated live vaccine, which is currently used in commercial poultry. Viruses that share the same recombination history are likely derived from the same parent virus suggesting that Conn/Conn46/91 is reisolated Conn vaccine derived from the Conn/Conn46/66 virus. The FL/FL18288/71 virus also shares all 4 transferred fragments with the Conn viruses, however; FL/FL18288/71 and Conn viruses are different serotypes suggesting that FL/FL18288/71 is a field virus that emerged due to point mutations accumulating in spike over time rather than from recombination.
All 6 of the transferred fragments in CK/CH/ZJ971/97 are identical to all 6 of the transferred fragments in vaccine strain Mass/H120, providing compelling evidence that CK/CH/ZJ971/97 is reisolated Mass/H120 vaccine. That observation was also reported by Zhang et al. [23]. It is interesting that Mass/H52 (8 transferred fragments) and Mass/H120 (6 transferred fragments) share only 5 identical transferred fragments. The Mass/H52 and Mass/H120 viruses were isolated circa 1955 in the Netherlands and it is widely accepted that H stands for Holland, but it actually stands for Houben, the owner of the broiler farm where the viruses were isolated [24]. It is thought that Mass/H120 was derived from Mass/H52 but the actual relationship between the viruses is not certain. Our data indicates that they are not necessarily parent and progeny but they are closely related.
The Gray/Gray/60 and JMK/JMK/64 viruses share 99.7% nucleotide similarity across the entire genome and have 4 identical transferred fragments with JMK/JMK/64 having one additional fragment located in the 5’UTR, which is not found in Gray/Gray/60. It is well known that the Gray/Gray/60 virus is nephropathogenic, whereas the JMK/JMK/64 virus is strictly respirotropic. Perhaps sequence differences in the 5’UTR, which is involved in replication of the viral genome, play a role in the different pathobiologies observed for these viruses.
There is evidence that some transferred fragments in field viruses come from vaccines. As an example, CK/CH/LSD/051/06 has 3 of 7 and 2 of 7 transferred fragments in common with vaccine strains Mass/H52 and Mass/H120, respectively. In addition, the only fragments that USA viruses have in common with the viruses from China and Taiwan are fragments also associated with Mass type vaccines, which are used in both regions, providing further evidence that some of the fragments in field viruses come from vaccines. That result and the observation in Figure 1 that the viruses separated into clades based on geographic location also supports the conclusion that USA viruses have not recombined with Asian viruses.
A difference in the order of taxa in phylogenetic trees constructed from different regions of the genome is further evidence of recombination [25]. The ordering of taxa in sequential trees [26,27] was conducted and inconsistent phylogenetic relationships were observed for all of the examined virus strains across the entire genome, indicating a substantial amount of recombination (data not shown). There is a high number of breakpoints in the 1a region of the genome and immediately upstream of the S gene, which has been previously shown to be a ‘hot spot’ for recombination [9]. A phylogenetic compatibility matrix constructed at the 70% bootstrap level for 250 bp sequence fragments at 100 bp intervals also showed that recombination breakpoints were distributed throughout the IBV genomes (data not shown).
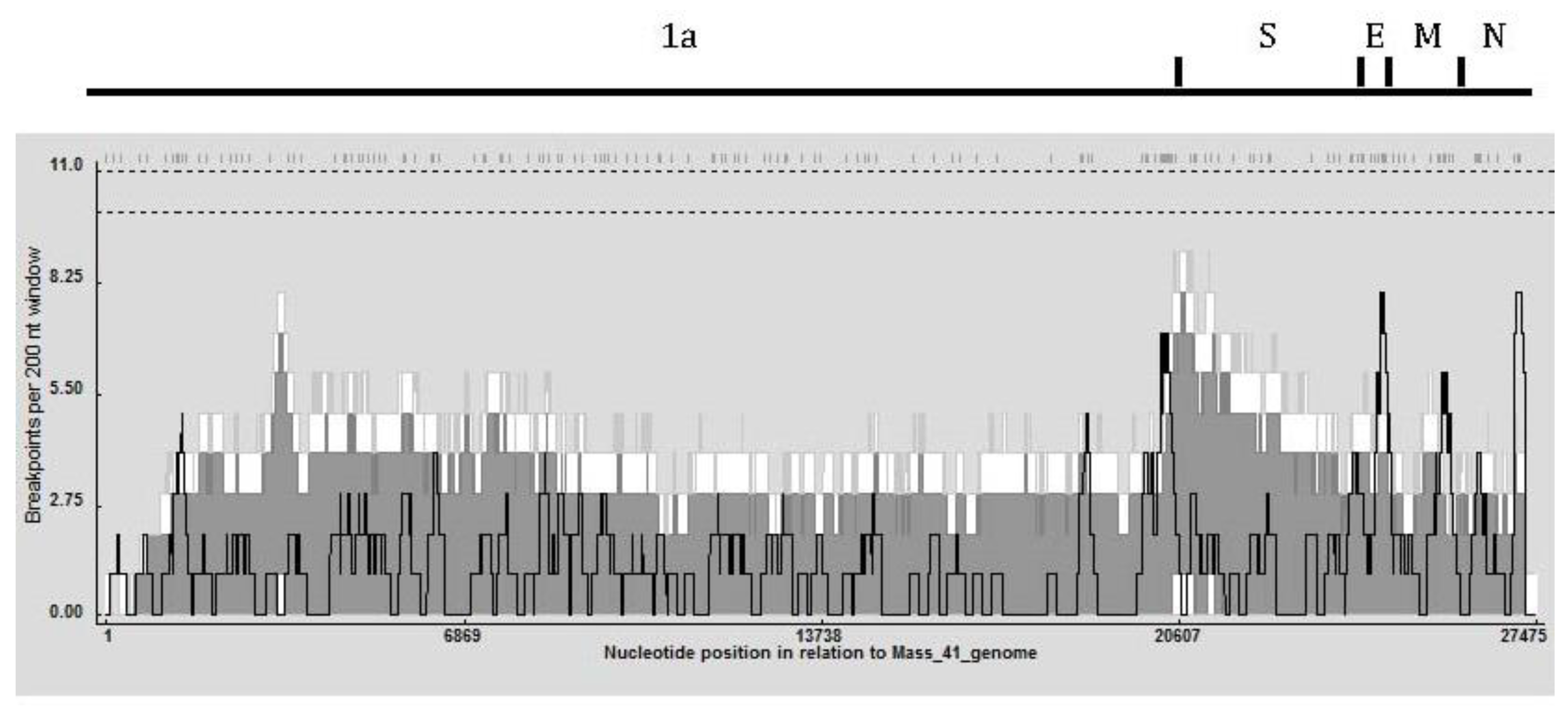
To determine recombination hot and cold spots, a recombination breakpoint distribution plot (Figure 3) was generated in RDP4 using a 200 nt window and 1,000 permutations [21]. No global hot-spot regions were observed in the 95% and 99% confidence thresholds (dotted lines at the top of the graph). The detectable recombination breakpoint positions are shown at the top of the figure and were distributed throughout the genome with a relatively high number clustered just upstream of the S gene. That region also had the highest breakpoint count within the 99% local hot/cold-spot confidence interval. A high number of breakpoints were also observed in the 1a region of the genome; nsp 2, nsp 3, and nsp 16, in the envelope and matrix protein genes and in a small area near the 3’UTR. Table 3 shows that nsp2, nsp3, nsp16 and spike genes were associated with the greatest number of transferred fragments, which is consistent with the location and number of breakpoints in Figure 3.
Recombination in the 1ab ORF area, which encodes the nonstructural proteins involved in the viral replication complex, has the potential to alter the pathogenicity of the virus [28]. The nsp 2 contains hydrophobic residues that likely anchor the replication complex to the Golgi [29]. The nsp 3 encodes the protease PLP2 which cleaves nsps 2, 3, and 4 and an area with ADP-ribose 1’-phosphatase (ADRP) activity. The protease PLP2 has been shown to have deubiquinating-like activity [30] and also to be a type I interferon (IFN) antagonist [31]. Changes in the amino acid composition of this area could affect the ability of the virus to replicate in a variety of cell types. The ADRP region of nsp 3 is conserved among coronaviruses [32,33], and a recent study suggested a biological role for the coronavirus ADRP in modulating the expression of pro-inflammatory immune modulators such as tumor necrosis factor alpha and interleukin-6 [34]. Recombination in this area could alter the pathogenicity of the virus by modulating host cytokine expression. The nsp16 is reported to be an S-adenosyl-L-methionine (AdoMet)-dependent RNA (nucleoside-2’O)-methyltransferase (2’O-MTase) responsible for capping the viral mRNA nascent transcripts [32]. An alteration in the efficiency of this protein could profoundly decrease not only viral replication but also pathogenicity. The spike glycoprotein of IBV on the surface of the virus plays a role in attachment to host cell receptors, membrane fusion and entry into the host cell. It also contains conformationally-dependent epitopes that induce virus-neutralizing and serotype specific antibodies [2,3]. We and others [6,7,8,10] have observed a relatively high number of breakpoints in and immediately upstream of spike, and changes to this region of the genome can result in the emergence of new genotypes and serotypes of IBV as well as new avian coronaviruses (i.e., TCoV). The envelope and matrix proteins are associated with virus assembly, and changes in those proteins could reduce the efficiency of virus particle formation and subsequent transmission of the virus. The 3’UTR is involved with binding of the viral RdRp and viral genome replication. Changes to the 3’UTR could affect replication efficiency and thus virulence of the virus.
3. Conclusions
In this study, evidence was obtained that recombination is occurring among avian coronavirus IBV isolates across their entire genome. Every sequence included in the analysis was recognized as a potential recipient of horizontally acquired sequences at some point in its viral evolutionary past. The nsp2, nsp3, nsp16 were associated with the greatest number of transferred fragments. In addition, the area immediately upstream of the spike gene had the highest number of recombination breakpoints. Breakpoints in the 1ab polyprotein gene have the potential to alter pathogenicity of the virus, and breakpoints near or in spike have the potential to lead to the emergence of new serotypes of IBV or new coronaviruses. Although the spike region determines the serotype of the virus, the remainder of the genome may be a mosaic of sequence fragments from a variety of gamma-coronaviruses. The only evidence of a gamma-coronavirus possibly recombining with an alpha or beta-coronavirus was the discovery of the mosaic nature of the SARS-coronavirus genome [35]. Although this type of recombination is possible it appears to be rare in nature.
In this study, we characterized recombination in the full-length genomes of avian gamma-coronavirus IBV strains from around the world. Our bioinformatic analysis was similar to a previous study on enteroviruses [36] and we found that recombination in IBV is more extensive than formerly thought, involving regions across the entire genome. Our data suggests that reticulate evolution due to a high frequency of recombination in IBV, likely plays a major role in the generation of new serotypes of the virus. The characterization, distribution and frequency of recombination breakpoints are important information that will further our understanding of the mechanisms behind the diversity and evolution of these viruses so that better control methods can be developed.
4. Materials and Methods
4.1. Viruses and Viral RNA Extraction
All of the viruses sequenced in this study (Table 4), were propagated in 9–11 day-old specific-pathogen-free (SPF) embryonated eggs as described [37]. Total RNA was isolated from 200 μL of allantoic fluid collected from the infected eggs using the High Pure RNA Isolation Kit (Roche Applied Science, Mannheim, Germany) following the manufacturer’s instructions.
4.2. RT-PCR Amplification and Sequencing
The amplification reactions were carried out using strand displacement RT-PCR or one step RT-PCR. Strand displacement RT-PCR uses a random (at the 3’ end) primer and an amplification primer. The sequence of the random primer was (AGCGGGGGTTGTCGAATGTTTGANNNN) and the sequence of the amplification primer was (AGCGGGGGTTGTCGAATGTTTGA). The RT-PCR reaction was carried out using the TaKaRa RNA LA PCR kit (Takara Bio. Inc., Otsu, Shiga, Japan) according to the manufacturer’s protocol. A DNA Engine Peltier thermocycler (Bio-Rad Laboratories Inc., Hercules, CA, USA) was used for the RT reaction, which included an RNA denaturing step at 65 °C for 10 min followed by 30 °C for 10 min, 42 °C for 60 min, 99 °C for 5 min, and 5 °C for 5 min. The PCR reaction was run on the same machine as the RT step and included a one-time initial denaturation step of 94 °C for 2 min, followed by 30 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 3 min.
The PCR products were agarose gel purified using the QIAquick gel extraction kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. The PCR products were cloned into the TOPOXL vector using the TOPOXL cloning kit (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s protocol to prepare cDNA libraries for sequencing.
Plasmid DNA from the libraries of the cloned cDNA fragments for each virus was isolated using an alkaline lysis method modified for the 96-well format and incorporating both Hydra and Tomtek robots. Sequencing reactions were performed using the BigDye™ Terminator® Cycle Sequencing Kit Version 3.1 (Applied Biosystems, Foster City, CA, USA) and MJ Research (Watertown, MA, USA) thermocyclers. Sephadex filter plates were used to filter each reaction into Perkin-Elmer MicroAmp Optical 96-well plates. A 1/12-strength sequencing reaction on an ABI 3730 was used to sequence each clone from both the 5’ and 3’ ends.
Primers for one-step RT-PCR were specifically designed for each virus (Supplemental Table 2). Viral RNA was amplified using the Titan One Tube RT-PCR kit (Roche Diagnostics, Indianapolis, IN, USA) following manufacturer’s instructions. A DNA Engine Peltier Thermocycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used for the RT-PCR reaction, which had the following steps: one cycle of 42 °C for 60 min and 95 °C for 5 min, followed by 10 cycles of 94 °C for 30 s, 50 °C for 30 s, and 68 °C for 1 min 30 s, and then 25 cycles of 94 °C for 30 s, 50 °C for 30 s, 68 °C for 1 min and 30 s adding 5 s with each cycle.
The resulting PCR products were agarose gel purified using the QIAquick gel extraction kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. The resulting cDNA was sequenced using ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s protocol. The reactions were prepared for sequencing by centrifugation through either a Centri-Sep column (Applied Biosystems, Foster City, CA, USA) or using the Edge system (EdgeBio, Gaithersburg, MD, USA) plate. The samples were sequenced at the Georgia Genomics Facility (University of Georgia, Athens, GA, USA).
4.3. Genome Assembly and Analysis
Chromatogram files and trace data were read and assembled using SeqMan Pro, and genome annotation was conducted with SeqBuilder (DNASTAR, Inc., Madison, WI, USA). Each sequence was aligned to a representative genome; Mass/Mass41/41 (GenBank accession #AY851295), or CAL99/CAL99/99 (GenBank accession #AY514485) as a backbone for genome assembly.
Whole genome analyses were generated and phylogenetic trees constructed with the Neighbor-Joining method with 1000 bootstrap replicates as well as with Minimum Evolution, Maximum Parsimony and UPGMA methods [17].
4.4. GenBank Accession Numbers
Virus genome sequences generated in this study were submitted to GenBank and assigned the following accession numbers: CAV/CAV56b/91 (GU393331), DE/DE072/92 (GU393332), FL/FL18288/71 (GU393333), Gray/Gray/60 (GU393334), Mass/H120 (GU393335), Holte/Holte/54 (GU393336), Iowa/Iowa97/56 (GU393337), JMK/JMK/64 (GU393338).
GenBank accession numbers for full-length sequences used as reference in this study are: Mass/Mass41/41 (AY851295), Mass/H52 (EU817497), Ark/Ark-DPI-p11/81 (EU418976), Ark-DPI-p101/91 (EU418975), CAV/CAV99/99 (AY514485), CK/CH/EP3 (DQ001338), CK/CH/p65 (DQ001339), Mass/Beaudette (NC_001451), NGA/A116E7/06 (FN430415), ITA/90254/05 (FN430414), TW/TW2575/98 (DQ646405), CK/CH/SC021202/02 (EU714029), CK/CH/ZJ971/97 (EU714028), CK/CH/BJ/97 (AY319651), CK/CH/SAIBK (DQ288927), CK/CH/LSD/051/06 (EU637854), CK/CH/DY07/07 (HM245923), CK/CH/CQ04-1/04 (HM245924), GA98/GA98/98 (GQ504723), PeafowlCcV/GD/KQ6/03 (AY641576), PartridgeCoV/GD/S14/03 (AY646283), TCoV/IN-540/94 (EU022525), TCoV/MN-ATCC (EU22526), TCoV/VA-74/03 (GQ427173), TCoV/TX-GL/01 (GQ427174), TCoV/IN-517/94 (GQ427175), TCoV/TX-1038/98 (GQ427176), TCoV/Canada-MG10 (EU095850) BulbulCoV/HKU11/09 (FJ376619), ThrushCoV/HKU12/09 (FJ376621), MuniaCoV/HKU13/09 (FJ376622), BelugaWhaleCoV/SW1/08 (NC_010646), FCoV/FIPV/WSU-79/1146 (DQ010921).
4.5. Detection of Networked Relationships and Recombination Break Points
We used Neighbor-net analysis to examine the IBV genomes for evidence of networked relationships and the pairwise homoplasy index (PHI) in SplitsTree (Version 4, Simmonics, University of Warwick, Coventry, UK) [20,38,39] to statistically determine the likelihood of recombination. In addition, the IBV genomes were examined for recombination breakpoints using the Recombination Detection Program (RDP4, Version 4, Simmonics, University of Warwick, Coventry, UK) [21,22]. Unless otherwise stated, default settings were used in all of the programs. The specific algorithms used were RDP [40], GENECONV [41], BOOTSCAN/RESCAN [40], MAXIMUM CHI SQUARE [42], CHIMAERA [43], SISCAN [44], and 3Seq [45]. We used more than one method to analyze the data because evaluation of these recombination detection methods using both simulated and empirical data showed that the results from only a single method were not very reliable [46]. Automasking was used for optimal recombination detection. The RDP analysis was run without a reference and a window size of 60, BOOTSCAN window size was increased to 500, MAXCHI and CHIMAERA number of variable sites per window was increased to 120, and the window size and step size for SISCAN was increased to 500 and 20, respectively. The window sizes were increased from their default settings because IBV has a high mutation rate, which can mask recombination signals. Increasing the window size was shown to increase the ratio of recombination signals relative to mutational “noise” [47].
4.6. Phylogenic Analysis of Sequential Genome Fragments
Inconsistent phylogenetic relationships between different regions of the viral genome provide further evidence of genetic recombination. Herein, we examined the order of avian gamma-coronavirus IBV strains in phylogenetic trees generated from sequential genome fragments using TreeOrder Scan (Version 1.6, Simmonics, University of Warwick, Coventry, UK) [26,27]. Changes in the tree position of taxa supported at the 70% or greater bootstrap level for a 250 bp sequence window were examined at 100 bp intervals. In addition, a phylogenetic compatibility matrix was constructed and used to examine the frequency and location of recombinations across the entire genome.
4.7. Recombination Site Detection
Potential recombination sites were identified using the RDP4 software [22] and a breakpoint map was constructed. A breakpoint density plot was then created from this map by moving a 200 nt window 1 nt at a time along the length of the map. The number of breakpoints falling within a window was plotted at the central window position. A 99% (upper) and 95% (lower) confidence threshold for globally significant breakpoint clusters (defined as windows with more breakpoint positions than the maximum found in >95% of the 1,000 permuted plots) was calculated. In addition, 99% and 95% confidence intervals were calculated for local breakpoint clusters (defined as windows with more breakpoint positions than the maximum found in >99% of the windows at that location in 1,000 permuted plots).
Acknowledgements
This work was supported by USDA, CSREES award number 2007-35600-17786. The authors would like to thank the technical help of Jon S. Robertson and Cornelia Lemke with sequencing.
References and Notes
- Lai, M.M.C.; Holmes, K.V. Coronaviridae: The viruses and their replication. In Fields Virology, 4th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 1, pp. 1163–1185. [Google Scholar]
- Cavanagh, D.; Mawditt, K.; Adzhar, A.; Gough, R.E.; Picault, J.P.; Naylor, C.J.; Haydon, D.; Shaw, K.; Britton, P. Does IBV change slowly despite the capacity of the spike protein to vary greatly? Adv. Exp. Med. Biol. 1998, 440, 729–734. [Google Scholar]
- Niesters, H.G.; Kusters, J.G.; Lenstra, J.A.; Spaan, W.J.; Horzined, M.C.; van der Zeijst, B.A. The neutralization epitopes on the spike protein of infectious bronchitis virus and their antigenic variation. Adv. Exp. Med. Biol. 1987, 218, 483–492. [Google Scholar]
- Holmes, E.C. The Evolution and Emergence of RNA Viruses, 1st ed.; Oxford University Press Inc.: New York, NY, USA, 2009. [Google Scholar]
- Lai, M. RNA Recombination in animal and plant viruses. Microbiol. Rev. 1992, 56, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; Jager, E.J.; Niesters, H.G.M.; van der Zeijst, B.A.M. Sequence evidence for RNA recombination in field isolates of avian coronavirus infectious bronchitis virus. Vaccine 1990, 8, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Karaca, K.; Parrish, C.R.; Naqi, S.A. A novel variant of avian infectious bronchitis virus resulting from recombination among three different strains. Arch. Virol. 1995, 140, 259–271. [Google Scholar] [CrossRef]
- Lee, C.W.; Jackwood, M.W. Origin and evolution of Georgia 98 (GA98), a new serotype of avian infectious bronchitis virus. Virus Res. 2001, 80, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Jackwood, M.W. Evidence of genetic diversity generated by recombination among avian coronavirus IBV. Arch. Virol. 2000, 145, 2135–48. [Google Scholar] [CrossRef]
- Estevez, C.; Villegas, P.; El-Attrache, J. A recombination event, induced in ovo, between a low passage infectious bronchitis virus field isolate and a highly embryo adaptedvaccine strain. Avian Dis. 2003, 47, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Mardani, K.; Noormohammadi, A.H.; Ignjatovic, J.; Browning, G.F. Naturally occurring recombination between distant strains of infectious bronchitis virus. Arch. Virol. 2010, 155, 1581–1586. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef]
- Decaro, N.; Mari, V.; Campolo, M.; Lorusso, A.; Camero, M.; Elia, G.; Martella, V.; Cordioli, P.; Enjuanes, L.; Buonavoglia, C. Recombinant canine coronaviruses related to transmissible gastroenteritis virus of Swine are circulating in dogs. J. Virol. 2009, 83, 1532–1537. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Boynton, T.O.; Hilt, D.A.; McKinley, E.T.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.; Lemke, C.; McCall, A.W.; Williams, S.M.; Jackwood, J.W.; Byrd, L.A. Emergence of a group 3 coronavirus through recombination. Virology 2010, 398, 98–108. [Google Scholar] [CrossRef]
- Lee, C.W.; Hilt, D.A.; Jackwood, M.W. Identification and analysis of the Georgia 98 serotype, a new serotype of infectious bronchitis virus. Avian Dis. 2001, 45, 164–172. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on 15 February 2011).
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.; Yuen, K.Y. Coronavirus genomics and bioinformatics analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Holmes, E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999, 80, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef]
- Heath, L.; van der Walt, E.; Varsani, A.; Martin, D.P. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef]
- Martin, D.P. Recombination detection and analysis using RDP3. Methods Mol. Biol. 2009, 537, 185–205. [Google Scholar]
- Zhang, Y.; Wang, H.-N.; Wang, T.; Fan, W.-Q.; Zhang, A.-Y.; Wei, K.; Tian, G.-B.; Yang, X. Complete genome sequence and recombination analysis of infectious bronchitis virus attenuated vaccine strain H120. Virus Genes 2010, 41, 377–388. [Google Scholar] [CrossRef]
- Hein, R. Poultry Health Consultant, Georgetown, DE, USA. Personal Communication, 2010. [Google Scholar]
- Woo, P.C.Y.; Lau, S.K.P.; Yip, C.C.Y.; Huang, Y.; Tsoi, H.-W.; Yuen, K-Y. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Welch, J. Frequency and dynamics of recombination within different species of human enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Midgley, S. Recombination in the genesis and evolution of hepatitis B virus genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef]
- Armesto, M.; Cavanagh, D.; Britton, P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS One 2009, 4, e7384. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Verheije, M.H.; Ulasli, M.; Shaltiel, I.A.; de Vries, L.A.; Reggiori, F.; Rottier, P.J.; de Haan, C.A. Dynamics of coronavirus replication-transcription complexes. J. Virol. 2010, 84, 2134–2149. [Google Scholar] [CrossRef]
- Lindner, H.A.; Fotouhi-Ardakani, N.; Lytvyn, V.; Lachance, P.; Sulea, T.; Menard, R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005, 79, 15199–15208. [Google Scholar] [CrossRef]
- Zheng, D.; Chen, G.; Guo, B.; Cheng, G.; Tang, H. PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 2008, 18, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Coronavirus genome: Prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic. Acids Res. 1989, 17, 4847–4861. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Lai, M.M. Putative papain-related thiol proteases of positive-strand RNA viruses. Identification of rubi- and aphthovirus proteases and delineation of a novel conserved domain associated with proteases of rubi-, alpha- and coronaviruses. FEBS Lett. 1991, 288, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.K.; Cervantes-Barragan, L.; Ludewig, B.; Thiel, V. Mouse hepatitis virus liver pathology is dependent on ADP-ribose-1’’-phosphatase, a viral function conserved in the alpha-like supergroup. J. Virol. 2008, 82, 12325–12334. [Google Scholar] [CrossRef]
- Zhang, X.W.; Yap, Y.L.; Danchin, A. Testing the hypothesis of a recombinant origin of the SARS-associated coronavirus. Arch. Virol. 2005, 150, 1–20. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Q.; Li, J.; Cao, W.; Zhang, J.X.; Zhang, L.; Zhang, W.; Shao, Z.J.; Yan, Y. Analysis of recombination and natural selection in human enterovirus 71. Virology 2010, 398, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Gelb, J.J.; Jackwood, M.W. Infectious Bronchitis. In A Laboratory Manual for the Isolation, Identification, and Characterization of Avian Pathogens, 5th ed.; Dufour-Zavala, L., Swayne, D.E., Glisson, J.R., Pearson, J.E., Reed, W.M., Jackwood, M.W., Woolcock, P., Eds.; American Association of Avian Pathologists: Kennett Square, PA, USA, 2008; pp. 146–149. [Google Scholar]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.; Moulton, V. Neighbor-net: An agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 13757–61372. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef]
- Posada, D. Evaluation of methods for detecting recombination from DNA sequences: Empirical data. Mol. Biol. Evol. 2002, 19, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Salminen, M.A.M. Detecting and characterising individual recombination events: practice. In The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing; Lemey, P., Salemi, M., Vandamme, A.M., Eds.; Cambridge University Press: Cambridge, UK, 2010; p. 723. [Google Scholar]
Figure 1.
Neighbor-joining method used to infer evolutionary history using full genomic sequence data available for the gamma-coronaviruses. The percentage of replicate trees in which the associated taxa clustered together in a bootstrap test of 1000 replicates is shown next to the branches. The p-distance scale is presented at the bottom of the figure. An asterisk (*) indicates a strain newly sequenced in this study. A number sign (#) indicates strains isolated in China that grouped with vaccine strains of infectious bronchitis virus (IBV).
Figure 1.
Neighbor-joining method used to infer evolutionary history using full genomic sequence data available for the gamma-coronaviruses. The percentage of replicate trees in which the associated taxa clustered together in a bootstrap test of 1000 replicates is shown next to the branches. The p-distance scale is presented at the bottom of the figure. An asterisk (*) indicates a strain newly sequenced in this study. A number sign (#) indicates strains isolated in China that grouped with vaccine strains of infectious bronchitis virus (IBV).

Figure 2.
Neighbor-net for the avian gamma-coronavirus IBV. The networked relationships are shown to indicate the presence of reticulate events. Boxes imply the likelihood of recombination.
Figure 2.
Neighbor-net for the avian gamma-coronavirus IBV. The networked relationships are shown to indicate the presence of reticulate events. Boxes imply the likelihood of recombination.

Figure 3.
Recombination breakpoint distribution plot generated for IBV using the Recombination Detection Program 4 (RDP4) showing the detectable recombination breakpoints. The plot was constructed using a 200 bp window moved 1 nucleotide at a time along the length of the genome. Recombination breakpoint positions are shown as hash marks at the top of the figure. The dashed lines under the breakpoint positions represent 99% (upper) and 95% (lower) confidence thresholds for globally significant breakpoint clusters (defined as windows with more breakpoint positions than the maximum found in >95% of the permuted plots). The dark gray and white areas are 95% confidence and 99% confidence intervals, respectively, for local breakpoint clusters (defined as windows with more breakpoint positions than the maximum found in >99% of the windows at that location in permuted plots), and the black line indicates the breakpoint count within the 200 bp window.
Figure 3.
Recombination breakpoint distribution plot generated for IBV using the Recombination Detection Program 4 (RDP4) showing the detectable recombination breakpoints. The plot was constructed using a 200 bp window moved 1 nucleotide at a time along the length of the genome. Recombination breakpoint positions are shown as hash marks at the top of the figure. The dashed lines under the breakpoint positions represent 99% (upper) and 95% (lower) confidence thresholds for globally significant breakpoint clusters (defined as windows with more breakpoint positions than the maximum found in >95% of the permuted plots). The dark gray and white areas are 95% confidence and 99% confidence intervals, respectively, for local breakpoint clusters (defined as windows with more breakpoint positions than the maximum found in >99% of the windows at that location in permuted plots), and the black line indicates the breakpoint count within the 200 bp window.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Genes and coding regions for eight strains of avian infectious bronchitis virus examined in this study.
Table 1.
Genes and coding regions for eight strains of avian infectious bronchitis virus examined in this study.
| ORFa | CAV/CAV56b/91 | DE/DE072/92 | FL/FL18288/71 | Gray/Gray/60 | Mass/H120 | Holte/Holte/54 | Iowa/Iowa97/56 | JMK/JMK/64 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Location | ntb | aac | Location | nt | aa | Location | nt | aa | Location | nt | aa | Location | nt | aa | Location | nt | aa | Location | nt | aa | Location | nt | aa | |
| 5’UTR | 1–527 | 527 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – | 1–528 | 528 | – |
| 1a | 528–12389 | 1,862 | 3953 | 529–12309 | 11781 | 3926 | 529–12387 | 11859 | 3952 | 529–12387 | 11859 | 3952 | 529–12330 | 11802 | 3933 | 529–12384 | 11856 | 3951 | 529–12390 | 11802 | 3933 | 529–12387 | 11859 | 3952 |
| 1ab | 528–20422 | 19895 | 6631 | 529–20336 | 19808 | 6602 | 529–20420 | 19892 | 6630 | 529–20420 | 19892 | 6630 | 529–20363 | 19835 | 6611 | 529–20414 | 19886 | 6628 | 529–20423 | 19895 | 6631 | 529–20421 | 19893 | 6630 |
| Spike | 20373–23873 | 3501 | 1166 | 20287–23739 | 3453 | 1150 | 20371–23838 | 3468 | 1155 | 20371–23874 | 3504 | 1167 | 20314–23802 | 3489 | 1162 | 20365–23871 | 3507 | 1168 | 20374–23880 | 3507 | 1168 | 20371–23877 | 3507 | 1168 |
| 3a | 23873–24046 | 174 | 57 | 23785–23958 | 174 | 57 | 23838–24011 | 164 | 54 | 23874–24047 | 174 | 57 | 23802–23975 | 174 | 57 | 23871–24044 | 174 | 57 | 23880–24053 | 174 | 57 | 23877–24050 | 174 | 57 |
| 3b | 24046–24240 | 195 | 64 | 23958–24152 | 195 | 64 | 24011–24202 | 192 | 63 | 24047–24241 | 195 | 64 | 23975–24169 | 195 | 64 | 24044–24238 | 195 | 64 | 24053–24247 | 195 | 64 | 24050–24244 | 195 | 64 |
| Envelope | 24221–24502 | 282 | 93 | 24133–24462 | 330 | 109 | 24186–24488 | 303 | 100 | 24222–24545 | 324 | 107 | 24150–24479 | 330 | 109 | 24219–24542 | 324 | 107 | 24228–24551 | 324 | 107 | 24225–24548 | 324 | 107 |
| Membrane | 24651–25175 | 525 | 174 | 24434–25111 | 678 | 225 | 24488–25156 | 669 | 222 | 24523–25188 | 666 | 221 | 24451–25128 | 678 | 225 | 24520–25188 | 667 | 222 | 24529–25140 | 612 | 203 | 24526–25197 | 672 | 223 |
| 4b | 25176–25460 | 285 | 94 | 25112–25396 | 285 | 94 | 25157–25441 | 285 | 94 | 25189–25428 | 240 | 79 | 25129–25371 | 243 | 80 | 25189–25473 | 285 | 94 | 25194–25478 | 285 | 94 | 25198–25329 | 132 | 43 |
| 4c | 25381–25554 | 174 | 57 | 25317–25487 | 171 | 56 | 25362–25532 | 171 | 56 | 25340–25510 | 171 | 56 | 25334–25504 | 171 | 56 | 25394–25534 | 141 | 46 | 25399–25539 | 141 | 46 | 25374–25568 | 195 | 64 |
| 5a | 25538–25735 | 198 | 65 | 25471–25668 | 198 | 65 | 25516–25713 | 198 | 65 | 25494–25691 | 198 | 65 | 25488–25685 | 198 | 65 | 25547–25744 | 198 | 65 | 25552–25749 | 198 | 65 | 25552–25749 | 198 | 65 |
| 5b | 25732–25980 | 249 | 82 | 25665–25913 | 249 | 82 | 25710–25958 | 249 | 82 | 25688–25936 | 249 | 82 | 25682–25930 | 249 | 82 | 25741–25989 | 249 | 82 | 25746–25994 | 249 | 82 | 25746–25994 | 249 | 82 |
| Nucleocapsid | 25923–27152 | 1230 | 409 | 25856–27085 | 1230 | 409 | 25901–27130 | 1230 | 409 | 25879–27111 | 1233 | 410 | 25873–27102 | 1230 | 409 | 25932–27161 | 1230 | 409 | 25937–27166 | 1230 | 409 | 25937–27166 | 1230 | 409 |
| 6b | 27161–27385 | 225 | 74 | 27094–27318 | 225 | 74 | 27139–27363 | 225 | 74 | – | – | – | 27126–27356 | 231 | 76 | – | – | – | 27175–27399 | 225 | 74 | 27175–27399 | 225 | 74 |
| 3’UTR | 27386–27663 | 248 | – | 27319–27591 | 273 | – | 27364–27616 | 253 | – | 27112–27568 | 455 | – | 27357–27632 | 276 | – | 27162–27246 | 85 | – | 27340–27662 | 323 | – | 27400–27793 | 393 | – |
a ORF = open reading frame; b nt = nucleotide; c aa = amino acid.
Table 2.
Recombination breakpoints a, genes and major and minor related sequences in other infectious bronchitis virus (IBV) strains.
Table 2.
Recombination breakpoints a, genes and major and minor related sequences in other infectious bronchitis virus (IBV) strains.
| Recombinant | Breakpoints | Genes b | Major Sequence c | Minor Sequence d | Detection Method | ||
|---|---|---|---|---|---|---|---|
| Begin | End | ||||||
| Ark/Ark-DPI-11/81 | 3,498 | 8,667 | 1ab (nsp 3, 4, and 5) | Conn/Conn46/66 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknowne (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,292 | 23,909 | 1ab (nsp16), Spike, 3a | Conn/Conn46/66 | Unknown (Mass/Mass41) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 21,613 | 23,856* | Spike, 3a | CAL/CAL99/99 | JMK/JMK/64 | RDP, Maxchi, Chimaera, SiSscan, 3Seq | ||
| Ark/Ark-DPI-101/81 | 3,498 | 8,667 | 1ab (nsp 3, 4, and 5) | Conn/Conn46/66 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,292 | 23,909 | 1ab (nsp16), Spike, 3a | Conn/Conn46/66 | Unknown (Mass/Mass41) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 21,613 | 23,856* | Spike, 3a | CAL/CAL99/99 | JMK/JMK/64 | RDP, Maxchi, Chimaera, SiSscan, 3Seq | ||
| CAL/CAL99/99 | 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 2,382 | 4,255* | 1ab (nsp2,nsp3) | DE/DE072/92 | Conn/Conn46/66 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 8,104 | 10,649* | 1ab (nsp 4, 5, and 6) | DE/DE072/92 | Conn/Conn46/66 | RDP, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,587* | 25,773 | Envelope, Membrane, 4b, 4c, 5a, 5b | Unknown (GA/GA98/98) | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| CAV/56b/91 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,392* | 4,558 | 1ab (nsp3) | Ark/Ark-DPI/81 | Conn/Conn46/91 | GENECONV, Maxchi, Chimaera, 3Seq | ||
| 8,104 | 10,649* | 1ab (nsp 4, 5, and 6) | DE/DE072/92 | Conn/Conn46/66 | RDP, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,292 | 23,909 | 1ab (nsp16), Spike, 3a | Conn/Conn46/66 | Unknown (Mass/Mass41) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,556 | 25,748 | Envelope, Membrane, 4b, 4c, 5a, 5b | Ark/Ark-DPI/81 | Unknown (CAL/CAL99/99) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| CK/CH/BJ/97 | 31* | 5,600 | 5’UTR, 1ab (nsp 2 and 3) | CK/CH/SAIBK | Unknown (CK/CH/CQ041/04) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| CK/CH/CQ04-1/04 | 60* | 4,711 | 5’UTR, 1ab (nsp 2 and 3) | CK/CH/SC021202/02 | CK/CH/DY-07/07 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 8,751 | 9,018 | 1 ab (nsp 5) | CK/CH/SC021202/02 | CK/CH/DY-07/07 | RDP, GENECONV, Maxchi, Chimaera | ||
| 9,626 | 18,737 | 1ab (nsp 5, 6, 7, 8, 9, 10, 11/12, 13, 14, 15) | CK/CH/SAIBK | CK/CH/DY-07/07 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 18,738* | 20,350 | 1ab (nsp 15 and 16) | CK/CH/SAIBK | ITA/90254/2005 | RDP, GENECONV, Maxchi, Chimaera | ||
| 20,160 | 21,138 | 1ab (nsp 16), Spike | JMK/JMK/64 | CK/CH/BJ/97 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| 27,120 | 27,354 | Nucleocapsid, 6b | JMK/JMK/64 | CK/CH/DY-07/07 | GENECONV, Maxchi, Chimaera, SiSscan | ||
| CK/CH/DY-07/07 | 1,170 | 5,017 | 1ab (nsp 2 and 3) | DE/DE072/92 | CK/CH/SAIBK | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 22,216 | 23,963 | Spike, 3a | CK/CH/BJ/97 | CK/CH/CQ04-1/04 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,455 | 25,662 | 4c, 5a | CK/CH/BJ/97 | CK/CH/CQ04-1/04 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| CK/CH/LSD/051/06 | 306 | 3,628* | 5’UTR, 1ab (nsp 2 and 3) | Mass/Mass41 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 1,453 | 2,743 | 1ab (nsp 2 and 3) | Mass/H52 | Mass/Mass41/41 | GENECONV, Maxchi, Chimaera, 3Seq | ||
| 13,668 | 14,734 | 1ab, (nsp 11/12) | Mass/Mass41/41 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 15,447 | 15,821 | 1ab (nsp 13) | Mass/Mass41/41 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,063 | 25,776 | Membrane, 4b, 4c, 5a, 5b | Unknown (Mass/Mass41/41) | Mass/H120 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,774* | 26,341 | 5b, Nucleocapsid | Mass/Mass41/41 | Mass/H120 | RDP, GENECONV, SiSscan, 3Seq | ||
| CK/CH/SAIBK | 7,241 | 9,126 | 1ab (nsp 3, 4,5) | CK/CH/SC0212/02 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 20,160 | 21,138 | 1ab (nsp 16), Spike | JMK/JMK/64 | CK/CH/BJ/97 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| CK/CH/SC021202/02 | 13,342 | 14,784 | 1ab (nsp 11/12) | CK/CH/SAIBK | CK/CH/DY-07/07 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 20,160 | 21,138 | 1ab (nsp 16), Spike | JMK/JMK/64 | CK/CH/BJ/97 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| 27,120 | 27,354 | Nucleocapsid, 6b | JMK/JMK/64 | CK/CH/DY-07/07 | GENECONV, Maxchi, Chimaera, SiSscan | ||
| CK/CH/ZJ971/97 | 0* | 11,115 | 5’UTR, 1ab (nsp 2, 3, 4, 5, 6, 7, and 8) | NGA/A116E7/06 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | |
| 306 | 3,628* | 5’UTR, 1ab (nsp 2 and 3) | Mass/Mass41 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 26,286 | 27,027 | Nucleocapsid, 6b, 3’UTR | Iowa/Iowa97/56 | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | ||
| 27,094 | 27,244 | Nucleocapsid, 6b | Iowa/Iowa97/56 | Unknown (TW/2575/98) | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| Conn/Conn46/66 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,361 | 21,981 | Spike | CAL/CAL99/99 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| Conn/Conn46/91 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,361 | 21,981 | Spike | CAL/CAL99/99 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| DE/DE072/92 | 0* | 11,115 | 5’UTR, 1ab (nsp 2, 3, 4, 5, 6, 7, and 8) | NGA/A116E7/06 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | |
| 18,776 | 19,911* | 1ab (nsp 15 and 16) | Mass/H120 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 19,934 | 24,431 | 1ab (nsp16), Spike, 3a, 3b, Envelope | Mass/H120 | Unknown (Mass/Mass41) | RDP, GENECOV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 23,504 | 24,431* | Spike, 3a, 3b, Envelope | CK/CH/CQ04-1/04 | CALCAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,575 | 27,482* | 5a, 5b, Nucleocapsid, 6b, 3’UTR | CK/CH/ZJ971/97 | JMK/JMK/64 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| FL/FL18288/71 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,361 | 21,981 | Spike | CAL/CAL99/99 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| GA98/0470/98 | 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 2,382 | 4,255* | 1ab, (nsp2 and 3) | DE/DE072/92 | Conn/Conn46/66 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 3,498 | 8,667 | 1ab (nsp 3, 4, and 5) | Conn/Conn46/66 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 9,569 | 9,770 | 1ab (nsp 5) | Gray/Gray/60 | Unknown (NGA/A116E7/06) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 13,072 | 20,186 | 1ab (nsp 11/12, 13, 14, 15, and 16) | Unknown (JMK/JMK/64) | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 23,504 | 24,431* | Spike, 3a, 3b, Envelope | CK/CH/CQ04-1/04 | CALCAL99/99 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,500* | 25,438 | Membrane, 4b | Conn/Conn46/66 | Mass/Mass41/41 | RDP, GENECONV, Chimaera, SiSscan, 3Seq | ||
| Gray/Gray/60 | 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 8,488 | 12,055 | 1ab (nsp 4, 5, 6, 7, 8, 9, and 10) | Unknown (CK/CH/LSD/051/06) | Conn/Conn46/91 | RDP, GENECONV, Maxchi, Chimaera | ||
| 13,070* | 14,216 | 1ab (nsp 11/12) | Unknown (CAV/56b/91) | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,131 | 27,145 | 3b, Envelope, Membrane, 4b, 4c, 5a, 5b, Nucleocapsid, 3’UTR | Ark/Ark-DPI/81 | Unknown (Conn/Conn46/91) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| Holte/Holte/54 | 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| Iowa/Iowa97/56 | 0* | 4,368 | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 4,368 | 5,144 | 1ab (nsp 3) | Holte/Holte/54 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| ITA/90254/05 | 16,367 | 25,699 | 1ab (nsp 13, 14, 15, 16) Spike, 3a, 3b, Envelope, Membrane, 4b, 4c, 5a | GA98/0470/98 | CK/CH/BJ/97 | RDP, GENCOV, Maxchi, SiSscan | |
| 22,216 | 23,963 | Spike, 3a | CK/CH/BJ/97 | CK/CH/CQ04-1/04 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,423 | 25,632* | Envelope, Membrane, 4b, 4c, 5a | CK/CH/DY-07/07 | NGA/A116E7/06 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | ||
| JMK/JMK/64 | 0* | 1,512 | 1ab (nsp 2) | ITA/90254/2005 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| 0* | 4,368* | 5’UTR,1ab (nsp 2 and 3) | Ark/Ark-DPI/81 | Unknown (DE/DE072/92) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 8,488 | 12,055 | 1ab (nsp 4, 5, 6, 7, 8, 9, and 10) | Unknown (CK/CH/LSD/051/06) | Conn/Conn46/91 | RDP, GENECONV, Maxchi, Chimaera | ||
| 13,070* | 14,216 | 1ab (nsp 11/12) | Unknown (CAV/56b/91) | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 24,131 | 27,145 | 3b, Envelope, Membrane, 4b, 4c, 5a, 5b, Nucleocapsid, 3’UTR | Ark/Ark-DPI/81 | Unknown (Conn/Conn46/91) | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| Mass/H52 | 306 | 3,628* | 5’UTR, 1ab (nsp 2 and 3) | Mass/Mass41 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | |
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 19,925 | 20,168* | 1ab (nsp 16) | Mass/Mass41/41 | Mass/H120 | GENECONV, Maxchi, Chimaera, SiSscan | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41/41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 25,063 | 25,776 | Membrane, 4b, 4c, 5a, 5b | Unknown (Mass/Mass41/41) | Mass/H120 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 26,286 | 27,027 | Nucleocapsid, 6b, 3’UTR | Iowa/Iowa97/56 | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | ||
| 26,372 | 27,526* | Nucleocapsid, 6b, 3’UTR | Unknown (DE/DE072/92) | Mass/H120 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 27,094 | 27,244 | Nucleocapsid, 6b | Iowa/Iowa97/56 | Unknown (TW/2575/98) | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| Mass/H120 | 0* | 11,115 | 5’UTR, 1ab (nsp 2, 3, 4, 5, 6, 7, and 8) | NGA/A116E7/06 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | |
| 306 | 3,628* | 5’UTR, 1ab (nsp 2 and 3) | Mass/Mass41 | Ark/Ark-DPI/81 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 4,312 | 10,590 | 1ab (nsp 3, 4, 5, and 6) | CK/CH/LSD/051 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 20,203 | 24,772 | 1ab (nsp 16), Spike, 3a, 3b, Envelope, Membrane | NGA/A116E7/06 | Mass/Mass41 | RDP, GENECONV, Maxchi, Chimaera, SiSscan, 3Seq | ||
| 26,286 | 27,027 | Nucleocapsid, 6b, 3’UTR | Iowa/Iowa97/56 | CAL/CAL99/99 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | ||
| 27,094 | 27,244 | Nucleocapsid, 6b | Iowa/Iowa97/56 | Unknown (TW/2575/98) | RDP, GENECONV, Maxchi, Chimaera, SiSscan | ||
| NGA/A116E7/06 | 7,035 | 8,271 | 1ab (nsp 3 and 4) | Holte/Holte/54 | DE/DE072/92 | RDP, GENECONV, Maxchi, Chimaera, 3Seq | |
| TW/2575/98 | 20,160 | 21,138 | 1ab (nsp 16), Spike | JMK/JMK/64 | CK/CH/BJ/97 | RDP, GENECONV, Maxchi, Chimaera, SiSscan | |
| 27,120 | 27,354 | Nucleocapsid, 6b | JMK/JMK/64 | CK/CH/DY-07/07 | GENECONV, Maxchi, Chimaera, SiSscan | ||
* The actual breakpoint position is undetermined. Most likely it was overprinted by a subsequent recombination event. a Only transferred gene fragments with statistical support of >1 × 10−12 (50 of 135 total unique fragments) are included in the table. b Genes indicate the coding sequences contained within the fragment introduced by recombination. c Major Sequence = Sequence most closely related to the sequence surrounding the transferred fragment. d Minor Sequence = Sequence closely related to the transferred fragment in the recombinant. e Unknown = only one parent and a recombinant need be in the alignment for a transferred fragment to be detectable. The sequence listed in parentheses was used to infer the existence of a missing parental sequence.
Table 3.
Number of transferred fragments associated with individual areas of the genome for all of the strains examined.
Table 3.
Number of transferred fragments associated with individual areas of the genome for all of the strains examined.
| Genomic Region | Number of Fragments a | % of Total |
|---|---|---|
| 5’UTR b | 8 | 4.2 |
| Nsp c 2 | 20 | 10.5 |
| nsp 3 | 33 | 17.3 |
| nsp 4 | 17 | 8.9 |
| nsp 5 | 15 | 7.9 |
| nsp6 | 10 | 8.3 |
| nsp 7 | 6 | 3.2 |
| nsp 8 | 6 | 3.2 |
| nsp9 | 4 | 2.1 |
| nsp 10 | 4 | 2.1 |
| nsp 11/12 | 13 | 6.8 |
| nsp 13 | 12 | 6.3 |
| nsp 14 | 10 | 5.3 |
| nsp 15 | 10 | 5.3 |
| nsp 16 | 19 | 10.0 |
| Spike | 30 | 15.8 |
| 3a | 14 | 7.4 |
| 3b | 13 | 6.8 |
| Envelope | 17 | 8.9 |
| Membrane | 17 | 8.9 |
| 4b | 12 | 6.3 |
| 4c | 12 | 6.3 |
| 5a | 15 | 7.9 |
| 5b | 11 | 5.8 |
| Nucleocapsid | 14 | 7.4 |
| 3’UTR | 13 | 6.8 |
a Genomic areas may be fully or only partially located in the transferred fragments. b UTR = untranslated region. c nsp = nonstructural protein.
Table 4.
Viruses sequenced in this study.
| Strain | Serotype | Origin | Source |
|---|---|---|---|
| CAV/CAV56b/91 | CAV | California, USA | P. Woolcock a |
| DE/DE072/92 | DE | Delmarva, USA | J. Gelb Jr b |
| FL/FL18288/71 | FL | Florida, USA | P. Villegas c |
| Gray/Gray/60 | Gray | Delmarva, USA | J. Gelb Jr. |
| Holte/Holte/54 | Holte | Wisconsin, USA | J. King d |
| Iowa/Iowa97/56 | Iowa | Iowa, USA | J. King |
| JMK/JMK/64 | JMK | Delmarva, USA | J. Gelb, Jr. |
| Mass/H120 | Mass | The Netherlands | J. King |
a University of California, Davis, CA, USA. b University of Delaware, Newark, DE, USA. c University of Georgia, Athens, GA, USA. d Southeast Poultry Research Laboratory, USDA/ARS, Athens, GA, USA.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus. Viruses 2011, 3, 1777-1799. https://doi.org/10.3390/v3091777
AMA Style
Thor SW, Hilt DA, Kissinger JC, Paterson AH, Jackwood MW. Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus. Viruses. 2011; 3(9):1777-1799. https://doi.org/10.3390/v3091777
Chicago/Turabian StyleThor, Sharmi W., Deborah A. Hilt, Jessica C. Kissinger, Andrew H. Paterson, and Mark W. Jackwood. 2011. "Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus" Viruses 3, no. 9: 1777-1799. https://doi.org/10.3390/v3091777