Hiding Lipid Presentation: Viral Interference with CD1d-Restricted Invariant Natural Killer T (iNKT) Cell Activation

{kind=link}
{kind=link}
Abstract
:1. Introduction

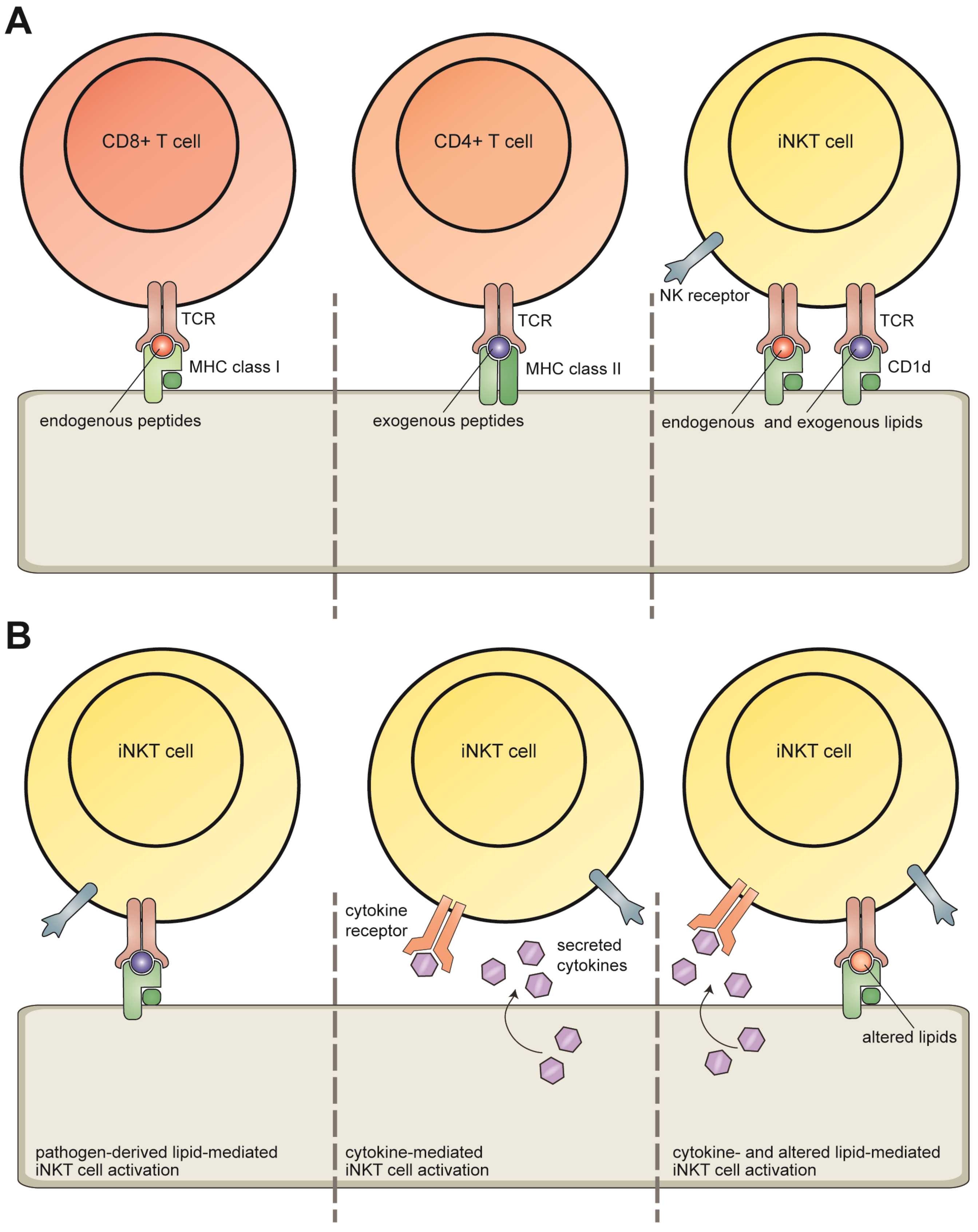
2. Invariant NKT Cells
3. CD1d Antigen Presentation
4. Lipid Antigens Presented by CD1d Molecules
4.1. Direct Recognition: Pathogen-Derived Lipids
4.2. Modulation of Self-Lipid Presentation during Viral Infection
5. iNKT Cells in Anti-Viral Defense
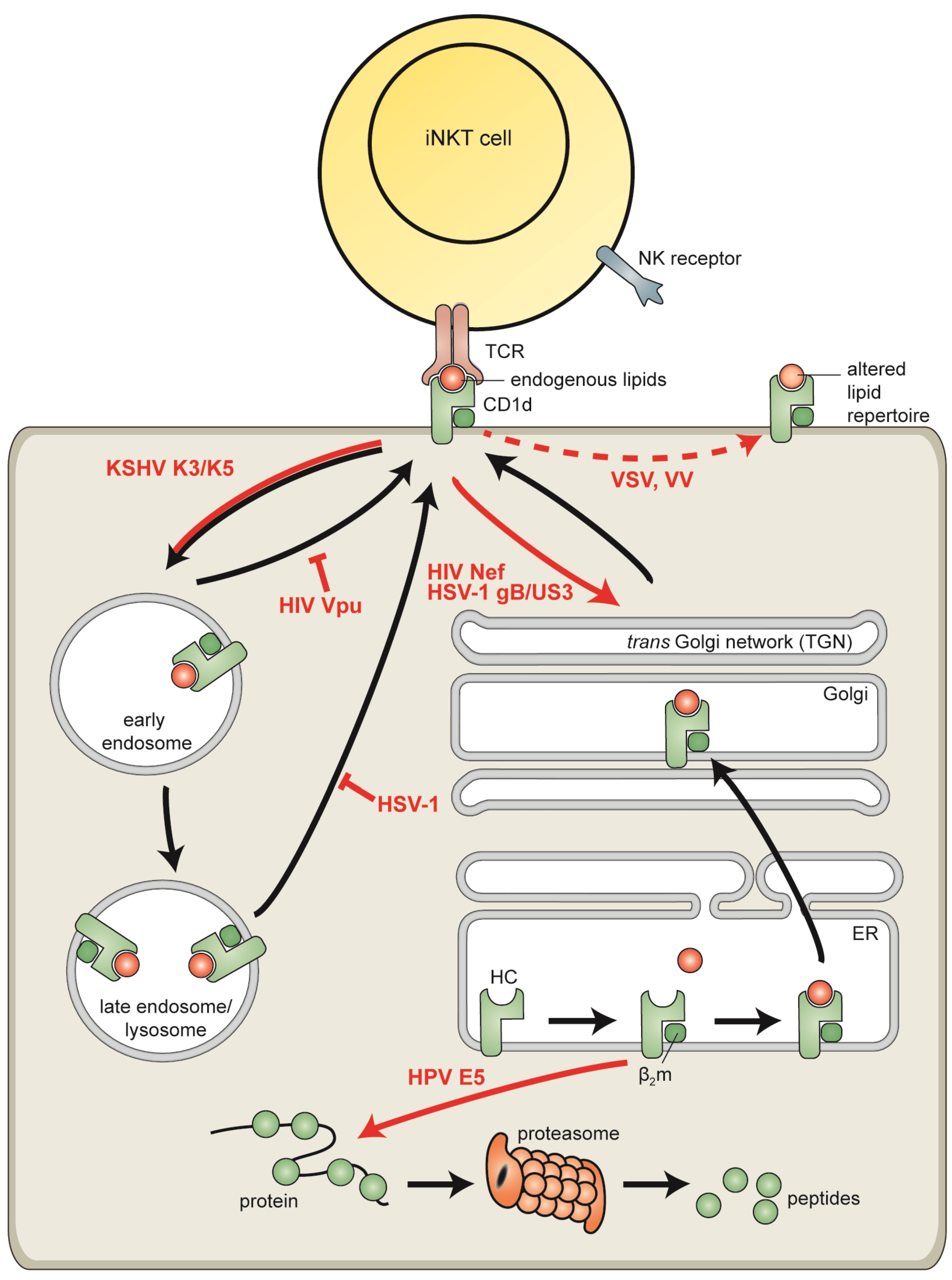
6. Viral Evasion of CD1d-Resticted Antigen Presentation

6.1. Human Immunodeficiency Virus
6.2. Vaccinia Virus and Vesicular Stomatitis Virus
6.3. Human Papillomaviruses
6.4. Kaposi’s Sarcoma-Associated Herpesvirus
6.5. Herpes Simplex Virus
7. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Hansen, T.H.; Bouvier, M. MHC class I antigen presentation: Learning from viral evasion strategies. Nat. Rev. Immunol. 2009, 9, 503–513. [Google Scholar] [CrossRef]
- Horst, D.; Verweij, M.C.; Davison, A.J.; Ressing, M.E.; Wiertz, E.J. Viral evasion of T cell immunity: Ancient mechanisms offering new applications. Curr. Opin. Immunol. 2011, 23, 96–103. [Google Scholar]
- Horst, D.; Ressing, M.E.; Wiertz, E.J. Exploiting human herpesvirus immune evasion for therapeutic gain: Potential and pitfalls. Immunol. Cell Biol. 2011, 89, 359–366. [Google Scholar] [CrossRef]
- Wiertz, E.J.; Devlin, R.; Collins, H.L.; Ressing, M.E. Herpesvirus interference with major histocompatibility complex class II-restricted T-cell activation. J. Virol. 2007, 81, 4389–4396. [Google Scholar] [CrossRef]
- Leslie, D.S.; Vincent, M.S.; Spada, F.M.; Das, H.; Sugita, M.; Morita, C.T.; Brenner, M.B. CD1-mediated gamma/delta T cell maturation of dendritic cells. J. Exp. Med. 2002, 196, 1575–1584. [Google Scholar] [CrossRef]
- Spada, F.M.; Grant, E.P.; Peters, P.J.; Sugita, M.; Melian, A.; Leslie, D.S.; Lee, H.K.; van Donselaar, .E.; Hanson, D.A.; Krensky, A.M.; et al. Self-recognition of CD1 by gamma/delta T cells: implications for innate immunity. J. Exp. Med. 2000, 191, 937–948. [Google Scholar]
- Godfrey, D.I.; Stankovic, S.; Baxter, A.G. Raising the NKT cell family. Nat. Immunol. 2010, 11, 197–206. [Google Scholar] [CrossRef]
- De Libero, G.; Mori, L. Mechanisms of lipid-antigen generation and presentation to T cells. Trends Immunol. 2006, 27, 485–492. [Google Scholar] [CrossRef]
- Chen, Y.H.; Wang, B.; Chun, T.; Zhao, L.; Cardell, S.; Behar, S.M.; Brenner, M.B.; Wang, C.R. Expression of CD1d2 on thymocytes is not sufficient for the development of NK T cells in CD1d1-deficient mice. J. Immunol. 1999, 162, 4560–4566. [Google Scholar]
- Rhost, S.; Sedimbi, S.; Kadri, N.; Cardell, S.L. Immunomodulatory type II natural killer T (NKT) lymphocytes in health and disease. Scand. J. Immunol. 2012, 76, 246–255. [Google Scholar] [CrossRef]
- Kuylenstierna, C.; Bjorkstrom, N.K.; Andersson, S.K.; Sahlstrom, P.; Bosnjak, L.; Paquin-Proulx, D.; Malmberg, K.J.; Ljunggren, H.G.; Moll, M.; Sandberg, J.K. NKG2D performs two functions in invariant NKT cells: direct TCR-independent activation of NK-like cytolysis and co-stimulation of activation by CD1d. Eur. J. Immunol. 2011, 41, 1913–1923. [Google Scholar] [CrossRef]
- Lee, H.H.; Meyer, E.H.; Goya, S.; Pichavant, M.; Kim, H.Y.; Bu, X.; Umetsu, S.E.; Jones, J.C.; Savage, P.B.; Iwakura, Y.; et al. Apoptotic cells activate NKT cells through T cell Ig-like mucin-like-1 resulting in airway hyperreactivity. J. Immunol. 2010, 185, 5225–5235. [Google Scholar]
- Brigl, M.; Bry, L.; Kent, S.C.; Gumperz, J.E.; Brenner, M.B. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat. Immunol. 2003, 4, 1230–1237. [Google Scholar] [CrossRef]
- Behar, S.M.; Dascher, C.C.; Grusby, M.J.; Wang, C.R.; Brenner, M.B. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. J. Exp. Med. 1999, 189, 1973–1980. [Google Scholar] [CrossRef]
- Sille, F.C.; Martin, C.; Jayaraman, P.; Rothchild, A.; Fortune, S.; Besra, G.S.; Behar, S.M.; Boes, M. Requirement for invariant chain in macrophages for Mycobacterium tuberculosis replication and CD1d antigen presentation. Infect. Immun. 2011, 79, 3053–3063. [Google Scholar] [CrossRef]
- Boes, M.; Stoppelenburg, A.J.; Sille, F.C. Endosomal processing for antigen presentation mediated by CD1 and Class I major histocompatibility complex: roads to display or destruction. Immunology 2009, 127, 163–170. [Google Scholar] [CrossRef]
- Yuan, W.; Qi, X.; Tsang, P.; Kang, S.J.; Illarionov, P.A.; Besra, G.S.; Gumperz, J.; Cresswell, P. Saposin B is the dominant saposin that facilitates lipid binding to human CD1d molecules. Proc. Natl. Acad. Sci. USA 2007, 104, 5551–5556. [Google Scholar]
- Brossay, L.; Jullien, D.; Cardell, S.; Sydora, B.C.; Burdin, N.; Modlin, R.L.; Kronenberg, M. Mouse CD1 is mainly expressed on hemopoietic-derived cells. J. Immunol. 1997, 159, 1216–1224. [Google Scholar]
- Amano, M.; Baumgarth, N.; Dick, M.D.; Brossay, L.; Kronenberg, M.; Herzenberg, L.A.; Strober, S. CD1 expression defines subsets of follicular and marginal zone B cells in the spleen: Beta 2-microglobulin-dependent and independent forms. J. Immunol. 1998, 161, 1710–1717. [Google Scholar]
- Liu, J.; Shaji, D.; Cho, S.; Du, W.; Gervay-Hague, J.; Brutkiewicz, R.R. A threonine-based targeting signal in the human CD1d cytoplasmic tail controls its functional expression. J. Immunol. 2010, 184, 4973–4981. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, W.; Veerapen, N.; Besra, G.; Cresswell, P. Calreticulin controls the rate of assembly of CD1d molecules in the endoplasmic reticulum. J. Biol. Chem. 2010, 285, 38283–38292. [Google Scholar]
- Sille, F.C.; Boxem, M.; Sprengers, D.; Veerapen, N.; Besra, G.; Boes, M. Distinct requirements for CD1d intracellular transport for development of V(alpha)14 iNKT cells. J. Immunol. 2009, 183, 1780–1788. [Google Scholar] [CrossRef]
- Lawton, A.P.; Prigozy, T.I.; Brossay, L.; Pei, B.; Khurana, A.; Martin, D.; Zhu, T.; Spate, K.; Ozga, M.; Honing, S.; et al. The mouse CD1d cytoplasmic tail mediates CD1d trafficking and antigen presentation by adaptor protein 3-dependent and -independent mechanisms. J. Immunol. 2005, 174, 3179–3186. [Google Scholar]
- Cernadas, M.; Sugita, M.; van der Wel, N.; Cao, X.; Gumperz, J.E.; Maltsev, S.; Besra, G.S.; Behar, S.M.; Peters, P.J.; Brenner, M.B. Lysosomal localization of murine CD1d mediated by AP-3 is necessary for NK T cell development. J. Immunol. 2003, 171, 4149–4155. [Google Scholar]
- Sugita, M.; Cernadas, M.; Brenner, M.B. New insights into pathways for CD1-mediated antigen presentation. Curr. Opin. Immunol. 2004, 16, 90–95. [Google Scholar]
- Sille, F.C.; Martin, C.; Jayaraman, P.; Rothchild, A.; Besra, G.S.; Behar, S.M.; Boes, M. Critical role for invariant chain in CD1d-mediated selection and maturation of Valpha14-invariant NKT cells. Immunol. Lett. 2011, 139, 33–41. [Google Scholar] [CrossRef]
- Jayawardena-Wolf, J.; Benlagha, K.; Chiu, Y.H.; Mehr, R.; Bendelac, A. CD1d endosomal trafficking is independently regulated by an intrinsic CD1d-encoded tyrosine motif and by the invariant chain. Immunity. 2001, 15, 897–908. [Google Scholar] [CrossRef]
- Kang, S.J.; Cresswell, P. Regulation of intracellular trafficking of human CD1d by association with MHC class II molecules. EMBO J. 2002, 21, 1650–1660. [Google Scholar] [CrossRef]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Motoki, K.; Ueno, H.; Nakagawa, R.; Sato, H.; Kondo, E.; et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 1997, 278, 1626–1629. [Google Scholar]
- Kobayashi, E.; Motoki, K.; Uchida, T.; Fukushima, H.; Koezuka, Y. KRN7000, a novel immunomodulator, and its antitumor activities. Oncol. Res. 1995, 7, 529–534. [Google Scholar]
- Cox, D.; Fox, L.; Tian, R.; Bardet, W.; Skaley, M.; Mojsilovic, D.; Gumperz, J.; Hildebrand, W. Determination of cellular lipids bound to human CD1d molecules. PLoS. One. 2009, 4, e5325. [Google Scholar]
- Kinjo, Y.; Wu, D.; Kim, G.; Xing, G.W.; Poles, M.A.; Ho, D.D.; Tsuji, M.; Kawahara, K.; Wong, C.H.; Kronenberg, M. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature 2005, 434, 520–525. [Google Scholar]
- Mattner, J.; Debord, K.L.; Ismail, N.; Goff, R.D.; Cantu, C., III; Zhou, D.; Saint-Mezard, P.; Wang, V.; Gao, Y.; Yin, N.; et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 2005, 434, 525–529. [Google Scholar]
- Sriram, V.; Du, W.; Gervay-Hague, J.; Brutkiewicz, R.R. Cell wall glycosphingolipids of Sphingomonas paucimobilis are CD1d-specific ligands for NKT cells. Eur. J. Immunol. 2005, 35, 1692–1701. [Google Scholar]
- Kinjo, Y.; Tupin, E.; Wu, D.; Fujio, M.; Garcia-Navarro, R.; Benhnia, M.R.; Zajonc, D.M.; Ben-Menachem, G.; Ainge, G.D.; Painter, G.F.; et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat. Immunol. 2006, 7, 978–986. [Google Scholar] [CrossRef]
- Kinjo, Y.; Illarionov, P.; Vela, J.L.; Pei, B.; Girardi, E.; Li, X.; Li, Y.; Imamura, M.; Kaneko, Y.; Okawara, A.; et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat. Immunol. 2011, 12, 966–974. [Google Scholar] [CrossRef]
- Chang, Y.J.; Kim, H.Y.; Albacker, L.A.; Lee, H.H.; Baumgarth, N.; Akira, S.; Savage, P.B.; Endo, S.; Yamamura, T.; Maaskant, J.; et al. Influenza infection in suckling mice expands an NKT cell subset that protects against airway hyperreactivity. J. Clin. Invest 2011, 121, 57–69. [Google Scholar]
- Zeissig, S.; Murata, K.; Sweet, L.; Publicover, J.; Hu, Z.; Kaser, A.; Bosse, E.; Iqbal, J.; Hussain, M.M.; Balschun, K.; et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat. Med. 2012, 18, 1060–1068. [Google Scholar]
- Bowie, A.G.; Haga, I.R. The role of Toll-like receptors in the host response to viruses. Mol. Immunol. 2005, 42, 859–867. [Google Scholar] [CrossRef]
- Brennan, K.; Bowie, A.G. Activation of host pattern recognition receptors by viruses. Curr. Opin. Microbiol. 2010, 13, 503–507. [Google Scholar]
- Paget, C.; Mallevaey, T.; Speak, A.O.; Torres, D.; Fontaine, J.; Sheehan, K.C.; Capron, M.; Ryffel, B.; Faveeuw, C.; Leite de, M.M.; et al. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity. 2007, 27, 597–609. [Google Scholar] [CrossRef]
- Salio, M.; Speak, A.O.; Shepherd, D.; Polzella, P.; Illarionov, P.A.; Veerapen, N.; Besra, G.S.; Platt, F.M.; Cerundolo, V. Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc. Natl. Acad. Sci. USA 2007, 104, 20490–20495. [Google Scholar]
- Muindi, K.; Cernadas, M.; Watts, G.F.; Royle, L.; Neville, D.C.; Dwek, R.A.; Besra, G.S.; Rudd, P.M.; Butters, T.D.; Brenner, M.B. Activation state and intracellular trafficking contribute to the repertoire of endogenous glycosphingolipids presented by CD1d. Proc. Natl. Acad. Sci. USA 2010, 107, 3052–3057. [Google Scholar]
- Brennan, P.J.; Tatituri, R.V.; Brigl, M.; Kim, E.Y.; Tuli, A.; Sanderson, J.P.; Gadola, S.D.; Hsu, F.F.; Besra, G.S.; Brenner, M.B. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat. Immunol. 2011, 12, 1202–1211. [Google Scholar] [CrossRef]
- Darmoise, A.; Teneberg, S.; Bouzonville, L.; Brady, R.O.; Beck, M.; Kaufmann, S.H.; Winau, F. Lysosomal alpha-galactosidase controls the generation of self lipid antigens for natural killer T cells. Immunity. 2010, 33, 216–228. [Google Scholar]
- Chen, X.; Wang, X.; Keaton, J.M.; Reddington, F.; Illarionov, P.A.; Besra, G.S.; Gumperz, J.E. Distinct endosomal trafficking requirements for presentation of autoantigens and exogenous lipids by human CD1d molecules. J. Immunol. 2007, 178, 6181–6190. [Google Scholar]
- Boes, M.; van der Wel, N.; Peperzak, V.; Kim, Y.M.; Peters, P.J.; Ploegh, H. In vivo control of endosomal architecture by class II-associated invariant chain and cathepsin S. Eur. J. Immunol. 2005, 35, 2552–2562. [Google Scholar] [CrossRef]
- Raftery, M.J.; Winau, F.; Giese, T.; Kaufmann, S.H.; Schaible, U.E.; Schonrich, G. Viral danger signals control CD1d de novo synthesis and NKT cell activation. Eur. J. Immunol. 2008, 38, 668–679. [Google Scholar] [CrossRef]
- Diana, J.; Lehuen, A. NKT cells: Friend or foe during viral infections? Eur. J. Immunol. 2009, 39, 3283–3291. [Google Scholar] [CrossRef]
- Tupin, E.; Kinjo, Y.; Kronenberg, M. The unique role of natural killer T cells in the response to microorganisms. Nat. Rev. Microbiol. 2007, 5, 405–417. [Google Scholar] [CrossRef]
- Levy, O.; Orange, J.S.; Hibberd, P.; Steinberg, S.; LaRussa, P.; Weinberg, A.; Wilson, S.B.; Shaulov, A.; Fleisher, G.; Geha, R.S.; et al. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. J. Infect. Dis. 2003, 188, 948–953. [Google Scholar] [CrossRef]
- Banovic, T.; Yanilla, M.; Simmons, R.; Robertson, I.; Schroder, W.A.; Raffelt, N.C.; Wilson, Y.A.; Hill, G.R.; Hogan, P.; Nourse, C.B. Disseminated varicella infection caused by varicella vaccine strain in a child with low invariant natural killer T cells and diminished CD1d expression. J. Infect. Dis. 2011, 204, 1893–1901. [Google Scholar]
- Nichols, K.E.; Hom, J.; Gong, S.Y.; Ganguly, A.; Ma, C.S.; Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L.; Koretzky, G.A.; Stein, P.L. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat. Med. 2005, 11, 340–345. [Google Scholar] [CrossRef]
- Pasquier, B.; Yin, L.; Fondaneche, M.C.; Relouzat, F.; Bloch-Queyrat, C.; Lambert, N.; Fischer, A.; de Saint-Basile, G.; Latour, S. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J. Exp. Med. 2005, 201, 695–701. [Google Scholar] [CrossRef]
- Rigaud, S.; Fondaneche, M.C.; Lambert, N.; Pasquier, B.; Mateo, V.; Soulas, P.; Galicier, L.; Le, D.F.; Rieux-Laucat, F.; Revy, P.; et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 2006, 444, 110–114. [Google Scholar]
- Felices, M.; Berg, L.J. The Tec kinases Itk and Rlk regulate NKT cell maturation, cytokine production, and survival. J. Immunol. 2008, 180, 3007–3018. [Google Scholar]
- Au-Yeung, B.B.; Fowell, D.J. A key role for Itk in both IFN gamma and IL-4 production by NKT cells. J. Immunol. 2007, 179, 111–119. [Google Scholar]
- Gadue, P.; Stein, P.L. NK T cell precursors exhibit differential cytokine regulation and require Itk for efficient maturation. J. Immunol. 2002, 169, 2397–2406. [Google Scholar]
- Huck, K.; Feyen, O.; Niehues, T.; Ruschendorf, F.; Hubner, N.; Laws, H.J.; Telieps, T.; Knapp, S.; Wacker, H.H.; Meindl, A.; et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J. Clin. Invest 2009, 119, 1350–1358. [Google Scholar] [CrossRef]
- Grubor-Bauk, B.; Simmons, A.; Mayrhofer, G.; Speck, P.G. Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NK[]T cells expressing the semivariant V alpha 14-J alpha 281 TCR. J. Immunol. 2003, 170, 1430–1434. [Google Scholar]
- Grubor-Bauk, B.; Arthur, J.L.; Mayrhofer, G. Importance of NKT cells in resistance to herpes simplex virus, fate of virus-infected neurons, and level of latency in mice. J. Virol. 2008, 82, 11073–11083. [Google Scholar] [CrossRef]
- Ashkar, A.A.; Rosenthal, K.L. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J. Virol. 2003, 77, 10168–10171. [Google Scholar] [CrossRef]
- Johnson, T.R.; Hong, S.; Van, K.L.; Koezuka, Y.; Graham, B.S. NK T cells contribute to expansion of CD8(+) T cells and amplification of antiviral immune responses to respiratory syncytial virus. J. Virol. 2002, 76, 4294–4303. [Google Scholar]
- Ishikawa, H.; Tanaka, K.; Kutsukake, E.; Fukui, T.; Sasaki, H.; Hata, A.; Noda, S.; Matsumoto, T. IFN-gamma production downstream of NKT cell activation in mice infected with influenza virus enhances the cytolytic activities of both NK cells and viral antigen-specific CD8+ T cells. Virology 2010, 407, 325–332. [Google Scholar] [CrossRef]
- van Dommelen, S.L.; Tabarias, H.A.; Smyth, M.J.; Gli-Esposti, M.A. Activation of natural killer (NK) T cells during murine cytomegalovirus infection enhances the antiviral response mediated by NK cells. J. Virol. 2003, 77, 1877–1884. [Google Scholar] [CrossRef]
- Ho, L.P.; Denney, L.; Luhn, K.; Teoh, D.; Clelland, C.; McMichael, A.J. Activation of invariant NKT cells enhances the innate immune response and improves the disease course in influenza A virus infection. Eur. J. Immunol. 2008, 38, 1913–1922. [Google Scholar] [CrossRef]
- Kakimi, K.; Guidotti, L.G.; Koezuka, Y.; Chisari, F.V. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp. Med. 2000, 192, 921–930. [Google Scholar] [CrossRef]
- Exley, M.A.; Bigley, N.J.; Cheng, O.; Tahir, S.M.; Smiley, S.T.; Carter, Q.L.; Stills, H.F.; Grusby, M.J.; Koezuka, Y.; Taniguchi, M.; et al. CD1d-reactive T-cell activation leads to amelioration of disease caused by diabetogenic encephalomyocarditis virus. J. Leukoc. Biol. 2001, 69, 713–718. [Google Scholar]
- Stumptner-Cuvelette, P.; Morchoisne, S.; Dugast, M.; Le Gall, S.; Raposo, G.; Schwartz, O.; Benaroch, P. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc. Natl. Acad. Sci. USA 2001, 98, 12144–12149. [Google Scholar]
- Li, D.; Xu, X.N. NKT cells in HIV-1 infection. Cell Res. 2008, 18, 817–822. [Google Scholar] [CrossRef]
- Hage, C.A.; Kohli, L.L.; Cho, S.; Brutkiewicz, R.R.; Twigg, H.L., III; Knox, K.S. Human immunodeficiency virus gp120 downregulates CD1d cell surface expression. Immunol. Lett. 2005, 98, 131–135. [Google Scholar] [CrossRef]
- Chen, N.; McCarthy, C.; Drakesmith, H.; Li, D.; Cerundolo, V.; McMichael, A.J.; Screaton, G.R.; Xu, X.N. HIV-1 down-regulates the expression of CD1d via Nef. Eur. J. Immunol. 2006, 36, 278–286. [Google Scholar] [CrossRef]
- Cho, S.; Knox, K.S.; Kohli, L.M.; He, J.J.; Exley, M.A.; Wilson, S.B.; Brutkiewicz, R.R. Impaired cell surface expression of human CD1d by the formation of an HIV-1 Nef/CD1d complex. Virology 2005, 337, 242–252. [Google Scholar] [CrossRef]
- Roeth, J.F.; Williams, M.; Kasper, M.R.; Filzen, T.M.; Collins, K.L. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J. Cell Biol. 2004, 167, 903–913. [Google Scholar] [CrossRef]
- Moll, M.; Andersson, S.K.; Smed-Sorensen, A.; Sandberg, J.K. Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood 2010, 116, 1876–1884. [Google Scholar] [CrossRef]
- Renukaradhya, G.J.; Webb, T.J.; Khan, M.A.; Lin, Y.L.; Du, W.; Gervay-Hague, J.; Brutkiewicz, R.R. Virus-induced inhibition of CD1d1-mediated antigen presentation: Reciprocal regulation by p38 and ERK. J. Immunol. 2005, 175, 4301–4308. [Google Scholar]
- Webb, T.J.; Litavecz, R.A.; Khan, M.A.; Du, W.; Gervay-Hague, J.; Renukaradhya, G.J.; Brutkiewicz, R.R. Inhibition of CD1d1-mediated antigen presentation by the vaccinia virus B1R and H5R molecules. Eur. J. Immunol. 2006, 36, 2595–2600. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O'Brien, P.M.; Campo, M.S. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Haghshenas, M.; Marchetti, B.; Campo, M.S. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int. J. Cancer 2006, 119, 2105–2112. [Google Scholar] [CrossRef]
- Zhang, B.; Li, P.; Wang, E.; Brahmi, Z.; Dunn, K.W.; Blum, J.S.; Roman, A. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virology 2003, 310, 100–108. [Google Scholar] [CrossRef]
- Miura, S.; Kawana, K.; Schust, D.J.; Fujii, T.; Yokoyama, T.; Iwasawa, Y.; Nagamatsu, T.; Adachi, K.; Tomio, A.; Tomio, K.; et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: A possible mechanism for immune evasion by HPV. J. Virol. 2010, 84, 11614–11623. [Google Scholar]
- Ganem, D. KSHV-induced oncogenesis. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, 2007; Chapter 56. [Google Scholar]
- Vossen, M.T.; Westerhout, E.M.; Soderberg-Naucler, C.; Wiertz, E.J. Viral immune evasion: A masterpiece of evolution. Immunogenetics 2002, 54, 527–542. [Google Scholar] [CrossRef]
- Griffin, B.D.; Verweij, M.C.; Wiertz, E.J. Herpesviruses and immunity: The art of evasion. Vet. Microbiol. 2010, 143, 89–100. [Google Scholar] [CrossRef]
- Lilley, B.N.; Ploegh, H.L. Viral modulation of antigen presentation: Manipulation of cellular targets in the ER and beyond. Immunol. Rev. 2005, 207, 126–144. [Google Scholar] [CrossRef]
- Wiertz, E.J.; Mukherjee, S.; Ploegh, H.L. Viruses use stealth technology to escape from the host immune system. Mol. Med Today 1997, 3, 116–123. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Hill, A.B. Viral interference with antigen presentation. Nat. Immunol. 2002, 3, 1019–1025. [Google Scholar] [CrossRef]
- Coscoy, L.; Ganem, D. Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. USA 2000, 97, 8051–8056. [Google Scholar] [CrossRef]
- Ishido, S.; Wang, C.; Lee, B.S.; Cohen, G.B.; Jung, J.U. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 2000, 74, 5300–5309. [Google Scholar]
- Ishido, S.; Choi, J.K.; Lee, B.S.; Wang, C.; DeMaria, M.; Johnson, R.P.; Cohen, G.B.; Jung, J.U. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity. 2000, 13, 365–374. [Google Scholar] [CrossRef]
- Coscoy, L.; Ganem, D. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Invest 2001, 107, 1599–1606. [Google Scholar] [CrossRef]
- Sanchez, D.J.; Gumperz, J.E.; Ganem, D. Regulation of CD1d expression and function by a herpesvirus infection. J. Clin. Invest 2005, 115, 1369–1378. [Google Scholar]
- Tigges, M.A.; Leng, S.; Johnson, D.C.; Burke, R.L. Human herpes simplex virus (HSV)-specific CD8+ CTL clones recognize HSV-2-infected fibroblasts after treatment with IFN-gamma or when virion host shutoff functions are disabled. J. Immunol. 1996, 156, 3901–3910. [Google Scholar]
- Yuan, W.; Dasgupta, A.; Cresswell, P. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat. Immunol. 2006, 7, 835–842. [Google Scholar] [CrossRef]
- Rao, P.; Pham, H.T.; Kulkarni, A.; Yang, Y.; Liu, X.; Knipe, D.M.; Cresswell, P.; Yuan, W. Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J. Virol. 2011, 85, 8093–8104. [Google Scholar]
- Temme, S.; Eis-Hubinger, A.M.; McLellan, A.D.; Koch, N. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J. Immunol. 2010, 184, 236–243. [Google Scholar]
- Neumann, J.; Eis-Hubinger, A.M.; Koch, N. Herpes simplex virus type 1 targets the MHC class II processing pathway for immune evasion. J. Immunol. 2003, 171, 3075–3083. [Google Scholar]
- Raftery, M.J.; Winau, F.; Kaufmann, S.H.; Schaible, U.E.; Schonrich, G. CD1 antigen presentation by human dendritic cells as a target for herpes simplex virus immune evasion. J. Immunol. 2006, 177, 6207–6214. [Google Scholar]
- Bosnjak, L.; Sahlstrom, P.; Paquin-Proulx, D.; Leeansyah, E.; Moll, M.; Sandberg, J.K. Contact-dependent interference with invariant NKT cell activation by herpes simplex virus-infected cells. J. Immunol. 2012, 188, 6216–6224. [Google Scholar]
- Zuo, J.; Thomas, W.; van Leeuwen, D.; Middeldorp, J.M.; Wiertz, E.J.; Ressing, M.E.; Rowe, M. The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J. Virol. 2008, 82, 2385–2393. [Google Scholar] [CrossRef]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar]
- Croft, N.P.; Shannon-Lowe, C.; Bell, A.I.; Horst, D.; Kremmer, E.; Ressing, M.E.; Wiertz, E.J.; Middeldorp, J.M.; Rowe, M.; Rickinson, A.B.; et al. Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS. Pathog. 2009, 5, e1000490. [Google Scholar] [CrossRef]
- Hislop, A.D.; Ressing, M.E.; van Leeuwen, D.; Pudney, V.A.; Horst, D.; Koppers-Lalic, D.; Croft, N.P.; Neefjes, J.J.; Rickinson, A.B.; Wiertz, E.J. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J. Exp. Med. 2007, 204, 1863–1873. [Google Scholar] [CrossRef]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.; Rowe, M. The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS. Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Quinn, L.L.; Tamblyn, J.; Thomas, W.A.; Feederle, R.; Delecluse, H.J.; Hislop, A.D.; Rowe, M. The Epstein-Barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J. Virol. 2011, 85, 1604–1614. [Google Scholar]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Gomez, R.; Heemskerk, B.; Toebes, M.; Mullen, M.M.; Jardetzky, T.S.; Longnecker, R.; Schilham, M.W.; et al. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. USA 2003, 100, 11583–11588. [Google Scholar]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Keating, S.; Gomez, R.; Franken, K.L.; Ottenhoff, T.H.; Spriggs, M.; Schumacher, T.N.; Hutt-Fletcher, L.M.; et al. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J. Virol. 2005, 79, 841–852. [Google Scholar]
- Trgovcich, J.; Johnson, D.; Roizman, B. Cell surface major histocompatibility complex class II proteins are regulated by the products of the gamma(1)34.5 and U(L)41 genes of herpes simplex virus 1. J. Virol. 2002, 76, 6974–6986. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Horst, D.; Geerdink, R.J.; Gram, A.M.; Stoppelenburg, A.J.; Ressing, M.E. Hiding Lipid Presentation: Viral Interference with CD1d-Restricted Invariant Natural Killer T (iNKT) Cell Activation. Viruses 2012, 4, 2379-2399. https://doi.org/10.3390/v4102379
Horst D, Geerdink RJ, Gram AM, Stoppelenburg AJ, Ressing ME. Hiding Lipid Presentation: Viral Interference with CD1d-Restricted Invariant Natural Killer T (iNKT) Cell Activation. Viruses. 2012; 4(10):2379-2399. https://doi.org/10.3390/v4102379
Chicago/Turabian StyleHorst, Daniëlle, Ruben J. Geerdink, Anna M. Gram, Arie J. Stoppelenburg, and Maaike E. Ressing. 2012. "Hiding Lipid Presentation: Viral Interference with CD1d-Restricted Invariant Natural Killer T (iNKT) Cell Activation" Viruses 4, no. 10: 2379-2399. https://doi.org/10.3390/v4102379