Advanced Vaccine Candidates for Lassa Fever

Department of Pharmacology and Toxicology, School of Medicine, and the Center for Predictive Medicine for Biodefense and Emerging Infectious Diseases, University of Louisville, Kentucky, USA
Viruses 2012, 4(11), 2514-2557; https://doi.org/10.3390/v4112514
Submission received: 27 September 2012
/
Revised: 20 October 2012
/
Accepted: 22 October 2012
/
Published: 29 October 2012
(This article belongs to the Special Issue Arenaviruses)
Abstract
:Lassa virus (LASV) is the most prominent human pathogen of the Arenaviridae. The virus is transmitted to humans by a rodent reservoir, Mastomys natalensis, and is capable of causing lethal Lassa Fever (LF). LASV has the highest human impact of any of the viral hemorrhagic fevers (with the exception of Dengue Fever) with an estimated several hundred thousand infections annually, resulting in thousands of deaths in Western Africa. The sizeable disease burden, numerous imported cases of LF in non-endemic countries, and the possibility that LASV can be used as an agent of biological warfare make a strong case for vaccine development. Presently there is no licensed vaccine against LF or approved treatment. Recently, several promising vaccine candidates have been developed which can potentially target different groups at risk. The purpose of this manuscript is to review the LASV pathogenesis and immune mechanisms involved in protection. The current status of pre-clinical development of the advanced vaccine candidates that have been tested in non-human primates will be discussed. Major scientific, manufacturing, and regulatory challenges will also be considered.
1. Introduction
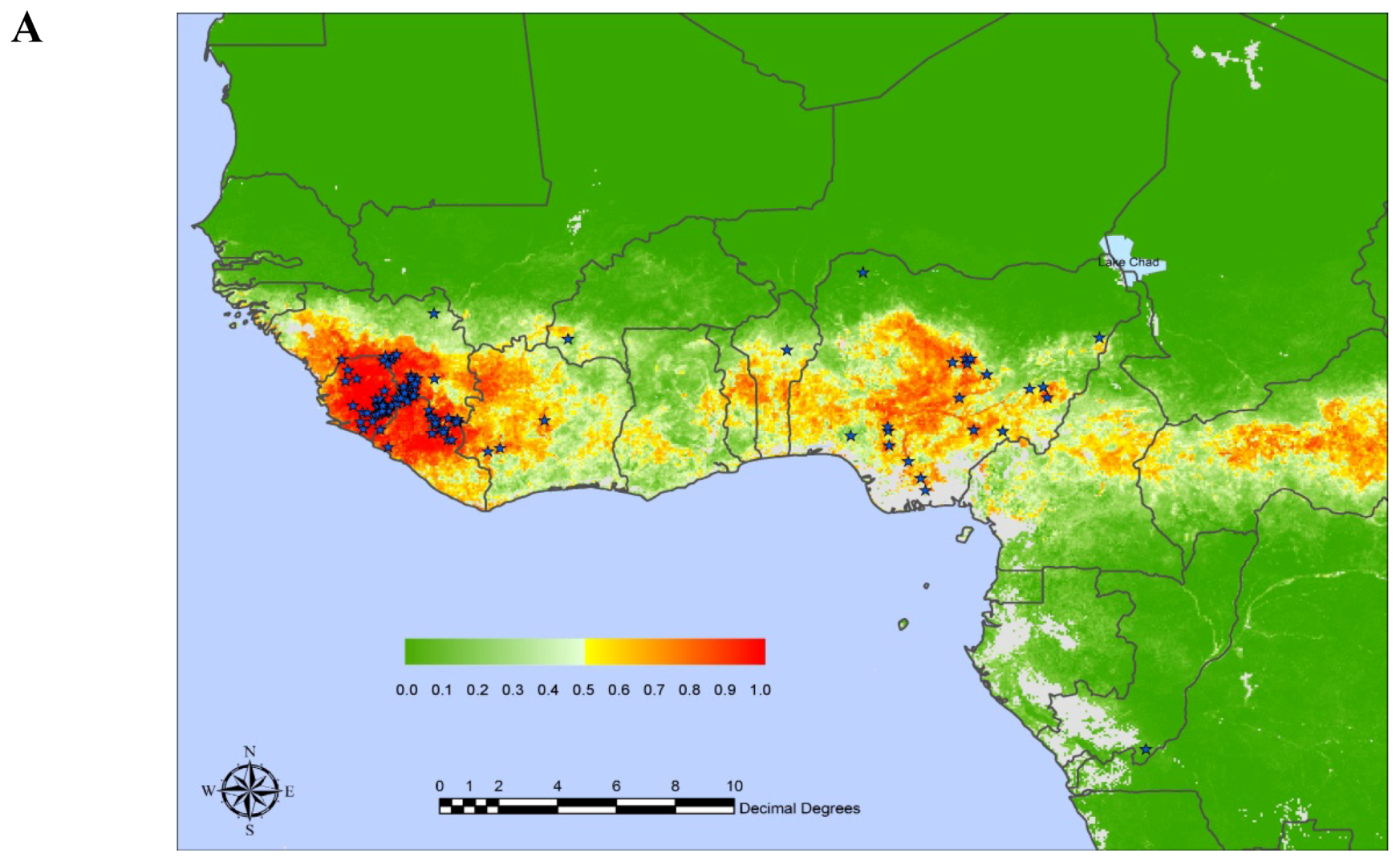
Lassa virus (LASV) is transmitted to humans by a rodent reservoir, Mastomys natalensis, and is capable of causing lethal Lassa Fever (LF) disease. There is no licensed vaccine for the prevention of LF and vaccine development efforts are hampered by both the high cost of non-human primate (NHP) animal models and biocontainment requirements (BSL-4) for study and development. LASV has the highest human impact of any of the hemorrhagic fever viruses (with the exception of Dengue fever) with an estimated 100,000-300,000 infections and 5,000-10,000 deaths annually in western Africa [1,2,3,4,5]. Based on prospective studies performed in four of the most affected countries, Guinea, Sierra-Leone, Liberia, and Nigeria, Richmond and Baglole [5] estimated that 59 million people are at risk of primary LASV infections with an annual incidence of disease as high as 3 million and as many as 67,000 deaths per year. The current LF predicted areas (Figure 1A) cover approximately 80% of Sierra-Leone and Liberia, 50% of Guinea, 40% of Nigeria, 30% of each of Côte d’Ivoire, Togo and Benin, and 10% of Ghana [3]. LASV was also detected in Mali [6] and LASV antibodies were detected in the Central African Republic, Democratic Republic of Congo, Senegal. Some experts believe that the population at risk includes most of the population of West Africa from Senegal to Nigeria and can be high as 200 million [5]. Recently performed genome-wide scans [7,8] suggest that LASV (and/or LASV-like viruses) may have been a driver of natural selection of genes implicated in LASV infectivity and immune responses in West African population.
Given the high annual incidence rate and mortality, it is arguable that LF is one of the most neglected tropical diseases, to the point that some have pointed out that if LF was a Developed World problem, there would be vociferous demands for control measures and vaccine [9]. An effective LASV vaccine is urgently needed not only for the general population, but also for healthcare and lab workers, as well as for military and other service personnel in West Africa. The vaccination strategies may differ for the various recipient populations. Whereas a multi-dose immunization regimen might be practical for medical providers and for military personnel, a single-dose vaccine would be ideal in endemic areas, where most of the target population is poor and live far from health care facilities [9].
Figure 1 (a).
Risk map of Lassa Fever in West Africa. Positive localities indicated by stars. The posterior probability color scale, from 0.0 (no risk) to 1.0 (highest risk) is shown as an inset. From E. Fichet-Calvet, D.J. Rogers [3] with permission.
Figure 1 (a).
Risk map of Lassa Fever in West Africa. Positive localities indicated by stars. The posterior probability color scale, from 0.0 (no risk) to 1.0 (highest risk) is shown as an inset. From E. Fichet-Calvet, D.J. Rogers [3] with permission.
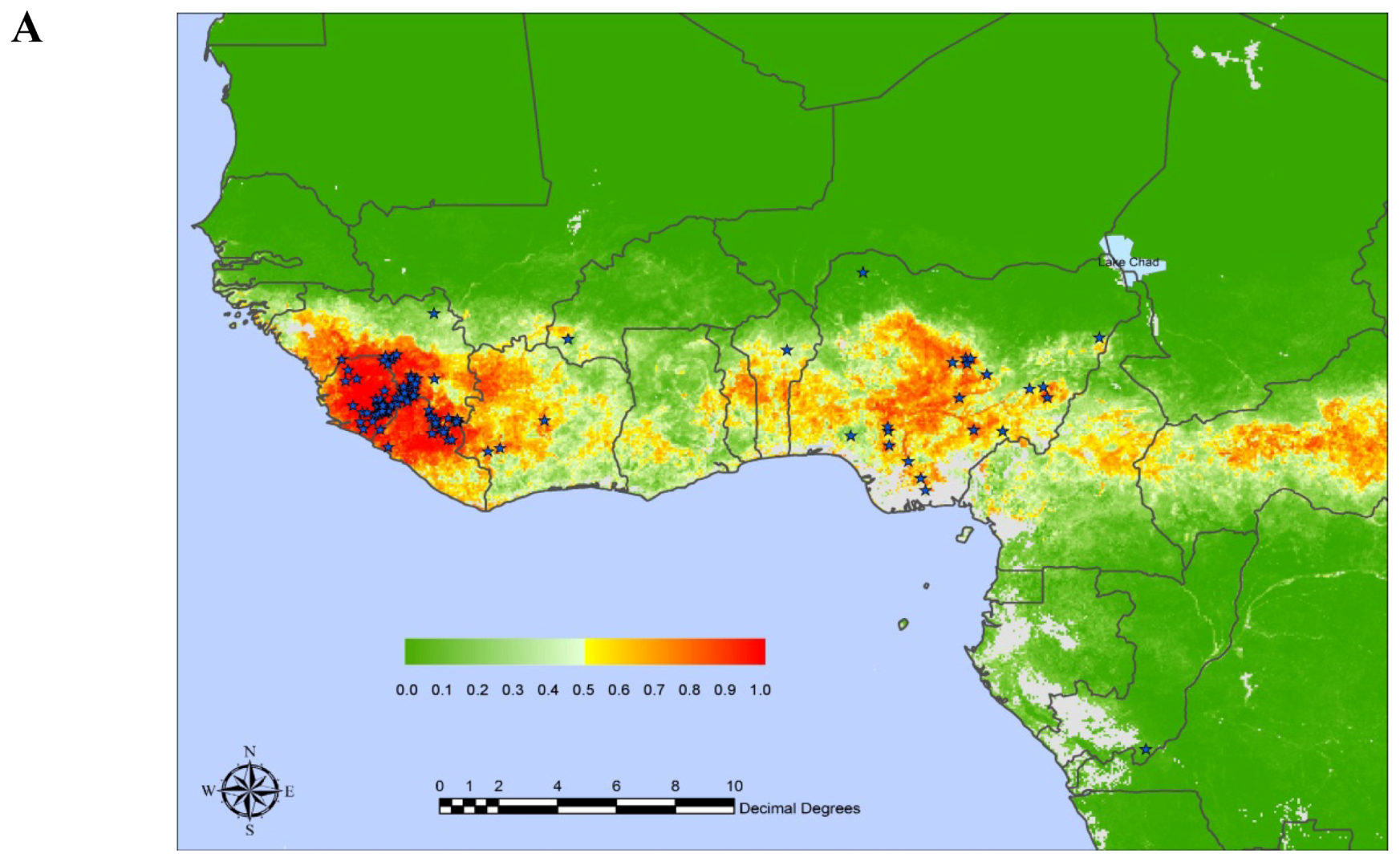
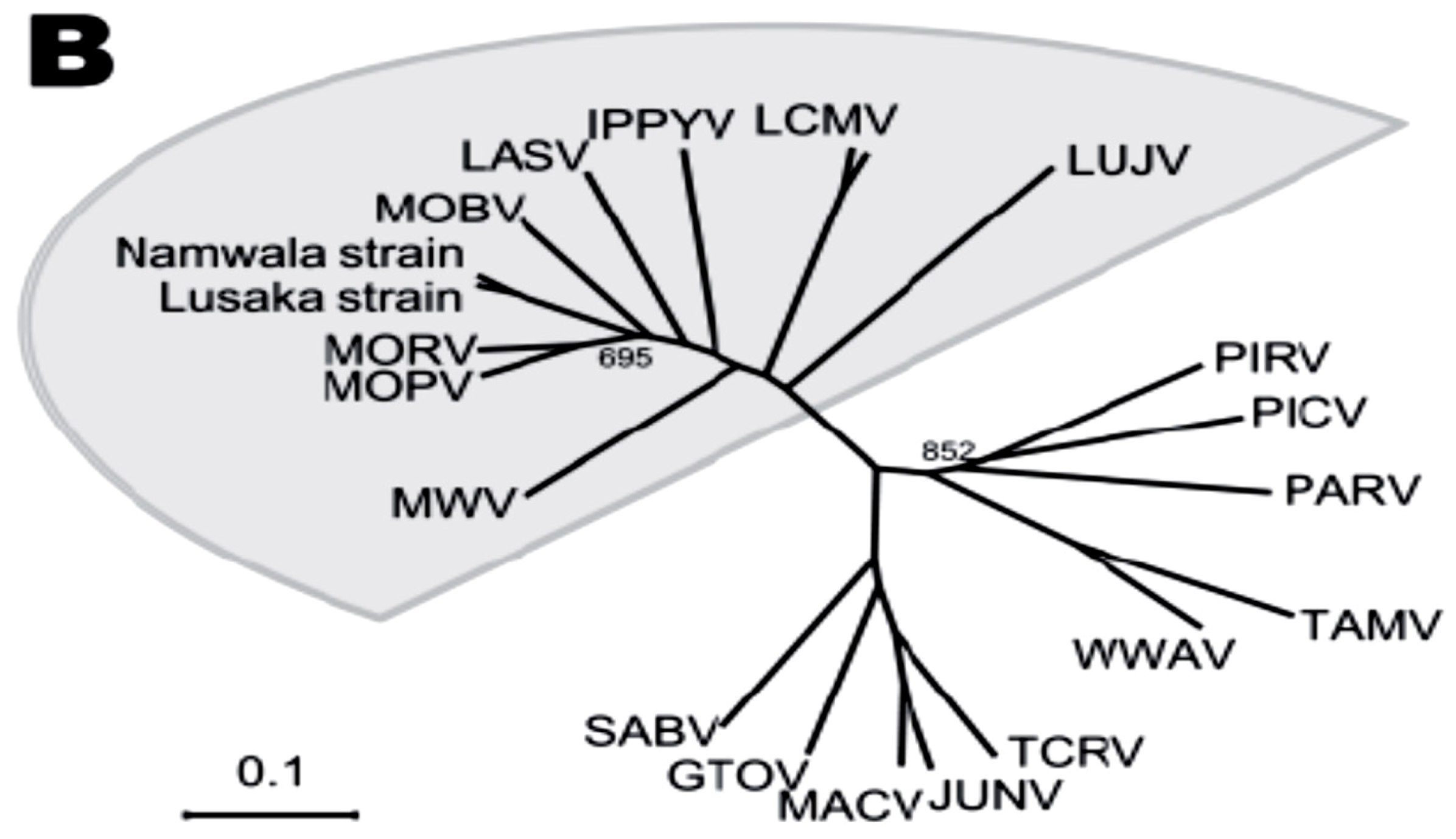
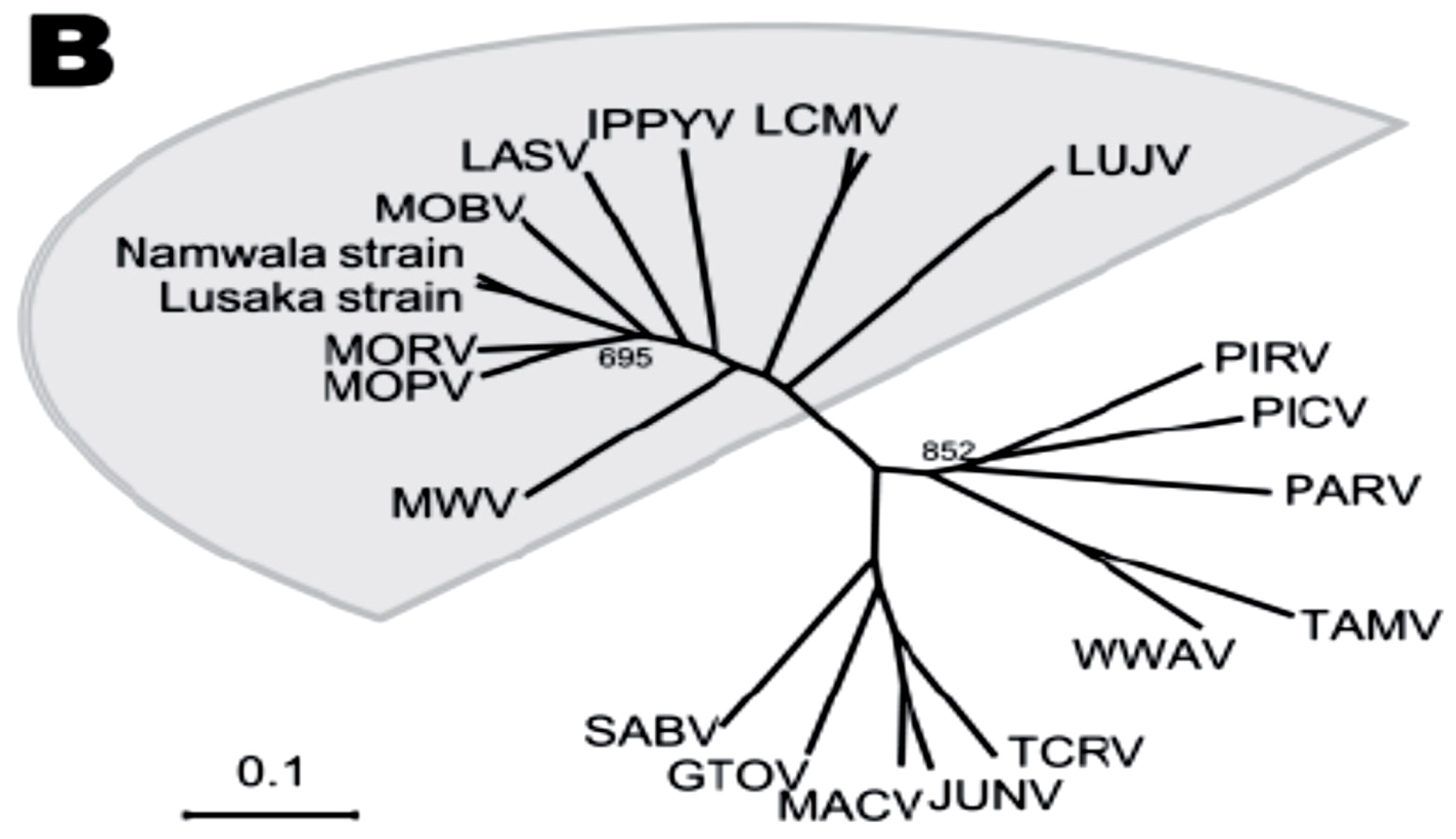
Figure 1 (b).
Phylogenetic relationships among the Old World arenaviruses (grey sector) based on analysis of the NP gene. From A. Ishii et al with permission [10]. See text for more details.
Figure 1 (b).
Phylogenetic relationships among the Old World arenaviruses (grey sector) based on analysis of the NP gene. From A. Ishii et al with permission [10]. See text for more details.
A cell-mediated immunity (CMI) plays the major role in the recovery and prevention (see below) and “a single shot of live attenuated candidate vaccine conferring life-long immunity is much preferred” [9]. In this review the current status of vaccine approaches to control LF will be overviewed with major attention to vaccine candidates tested in NHP models. Obstacles and challenges on the route of these vaccines into early stage of clinical development will be discussed as well.
3. Lassa Fever Pathogenesis and Mechanisms of Protection
As mentioned above, LASV is the most prevalent arenavirus in West Africa. Fortunately, ~ 80% of LASV-infected individual express sub-clinical or flu-like manifestations and the overall case-fatality rate is ~ 1-2%. However, in hospitalized patients and in some risk groups (pregnant women, children <5 years old, immunocompromised individuals, etc.) the fatality rate can be higher than 50% [1]. After recovery, in 29% of LF patients, acute manifested infection is accompanied by a sensorineural hearing deficit, which accounts for a permanent hearing loss in 18 % of survivors [50,51].
The pathogenesis of LF is still not clearly understood. Severe LASV infection is characterized by unchecked viremia, functional liver damage, and immunosuppression [1,52]. Host factors such as a cell receptor polymorphism [8], innate immunity [53,54,55,56], pro-inflammatory cyto/chemokines [56,57,58,59], adaptive CMI responses [60,61] as well as differences in pathogenic potential of LASV isolates [62] seem to play a role in outcome of LASV infection in humans. LASV replicates in target tissues (liver, spleen, lymph nodes, adrenal) without cytopathic effect, and the pathological damage to these tissues is usually not sufficient to implicate organ failure as the cause of death [63,64,65]. In fact, death from LF is caused mostly by uncontrolled sepsis-like terminal cardiogenic shock and internal bleeding. In contrast to HFs caused Ebola and Marburg viruses [66], a disseminated intravascular coagulation is not involved in LF pathogenesis, bleeding abnormalities are not common. Indeed, it has been even questioned if LF is pathologically a hemorrhagic fever [63,66].
Natural rodent-to-human LASV transmission occurs via inhalation of contaminated droplets/dust, or through ingestion of contaminated food [1,67]. Respiratory entry of arenaviruses occurs via basolateral receptors [68]. Recently we have shown that the viruses enter polarized Caco-2 cells from the basolateral site as well (Lukashevich et al; unpublished). These observations are in good confirmation with basolateral location of major LASV cell receptor, α-dystroglycan (αDG), and with general perception that basolateral entry provides easiest way to access target cells, macrophages and DCs. While entry requires the basolateral surface, LASV can be released from the apical cell surface. At least in polarized kidney-derived cells (MDCK), LASV virus-like particles (VLPs) exit cells from the apical site and this process was driven by GP [69]. Excretion of LASV from kidney of persistently infected M. natalensis is the major source of natural contamination for human exposure.
In spite of recent success in our understanding of LASV-host receptor interaction (rev. [70]), the detailed mechanism of LASV entry is unclear and the question remains how the virus crosses through respiratory and gastro-intestinal epithelia. LASV is remarkably resistant to low pH [71]. This factor combined with our epidemiological observations in Guinea, where hunting of peridomestic rodents and consumption of their meat are possible risk factors [67], suggest that gastric and/or intestinal epithelial cells can be the major gait entry. This hypothesis is supported by data from experimentally infected (through gastric rote) rhesus macaques [72]. The requirement of the virus for basolateral entry suggests that integrity of the epithelia must be compromised for the virus to get access to the basolateral site. However, as LASV infection is not cytopathic to epithelial [68,69] and endothelial [57] cells and does not appear to alter barrier function [69], external factors affecting epithelia integrity may contribute to the infection.
The virulence of LASV in experimentally infected animals is directly related to virus replication, and it is not related to classic T cell-mediated pathology (rev. [73,74]). Clinical studies showed that high viremia (≥ 1 x 103 TCID50/ml) was associated with high fatality rate and elevated viremia together with a high aspartate aminotransferase level (AST ≥ 150 IU/L) carried a risk of death in 78% of the cases. Similar observations were made in the LF model in NHP [27,72,74,75,76,77,78]. Based on data available so far, the major events leading to fatal outcome are summarized below: (i) from the initial sites of infection the virus spreads to regional lymph nodes, liver, spleen and infects tissue macrophages and DCs; (ii) release of cyto/chemokines from these cells recruits additional macrophages to sites of infection, amplifies deregulated host responses, contributes to loss of lymphocytes, down-regulates their functions; (iii) failure to properly initiate innate and adaptive immune response results in ineffective virus control; (iv) pro-inflammatory mediators from infected macrophages and DCs affect endothelial function and virus-induced impairment of vascular functions contributes to edema and shock at terminal stages; (v) infection of Kupffer cells (KCs), hepatocytes, and adrenal cortical cells additionally contributes to the hemodynamic and coagulation dysfunctions and results in hypotension and sodium loss with hypovolemia. Combined together, these events lead to sepsis-like shock leading to fatal outcome [2,52,66,79].
The production of pro-inflammatory cytokines results in the activation of antigen-presenting cells and T cell stimulation in the adaptive immune response and is required for clearance of viral infection. A large body of evidence accumulated during the last decade indicates that LASV-induced immunosuppression contributes to fatal outcome. We previously showed that replication of the OW pathogenic arenaviruses (LASV and LCMV-WE), but not non-pathogenic (MOPV and LCMV-ARM), was associated with the inhibition of pro-inflammatory cyto/chemokine responses in vitro and in vivo [57,80,81]. Indeed, the cytokine responses in LF patients and in experimentally infected NHP showed no evidence for a “cytokine storm”, which was observed in filovirus-induced hemorrhagic fevers (rev. [52]). In the progressed LF patients, fatal infection correlated with low or undetectable levels of pro-inflammatory cyto/chemokine [58]. At present, the immunosuppressive phenotype of fatal LASV infection is a well-accepted concept [52,82,83,84,85]. In human DCs-T-cell co-cultures, LASV induced only weak memory phenotype markers, while MOPV strongly stimulated CD8+ and CD4+ T cells, activation markers, proliferative responses, and CTL activities [83]. We have also shown recently that pathogenic arenaviruses, LASV and LCMV-WE, suppress NF-kB-mediated TLR2/Mal-dependent pro-inflammatory cytokine responses in infected monocytic cells [56]. In contrast, MOPV induced strong up-regulation of cytokines known to contribute to the development of robust adaptive immune responses. Similarly, in LCMV-ARM-infected mice, TLR2/MyD88/Mal signaling played an essential role in antiviral CD8+ T cell responses [86]. In the absence of MyD88, naive CD4+ T cells failed to differentiate into LCMV-specific CD4+ T cells [87]. Whether the blunted induction of cytokines is a direct or indirect outcome of virulent virus infection, the lack of innate stimulation likely positions the host to have a delayed and/or inefficient adaptive immune response to clear the infection. The immunosuppressive phenotype of LASV infection must be taken into consideration during rational vaccine design. An ideal vaccine has to induce strong, long-lasting protective immune responses prior LASV challenge.
The major cellular receptor for LASV, other OW arenaviruses, and Clade C of the NW viruses is α-DG, a cell surface receptor for extracellular matrix (ECM) proteins [88]. In the spleen, over 99% of α-DG is associated with DCs and less than 1% with CD4+ and CD8+ T cells. LASV and LCMV-WE54 (causes a LF-like fatal disease in NHPs) bind to α-DG with high affinity, preferentially infect DCs, effectively compete with and displace ECM molecules of DCs, and alter their ability to initiate an effective immune response [89,90]. In contrast, the low affinity of MOPV and LCMV-ARM (both viruses are not pathogenic for NHP) to α-DG correlates with their ability to induce TLR2/MyD88/Mal-dependent cytokine responses [56] and with the adaptive CMI responses to effectively control virus replication and restrict viral pathogenesis. However, several findings suggested that that α-DG is not the sole receptor for LASV and LCMV [89,91]. In fact, the level of α-DG is the highest in muscle tissues, where the LASV load in minimal. In contrast, the viral burden is very high in hepatocytes during the infection; these cells do not express α-DG. Recently two C-type lectin family members, DC-SIGN and LSECtin, and two TAM family members, Axl and Tyro3, have been recently identified as LASV and LCMV receptors [92,93], which may be responsible for replication in hepatocytes.
Recovery of LF patients and experimentally infected animals was not related to antibody levels and protection of animals in challenge experiments was associated with CMI [27,60,61,94,95,96]. Controlled clinical trials with human convalescent plasma containing high titers of antibodies failed to protect [74]. Although prime-boost immunization of primates with concentrated and gamma-irradiated LASV induced strong antibody responses to NP and GP, the immunized animals were not protected against challenge [97]. In naturally-infected individuals, anti-LASV antibodies had a short life-time and become undetectable in a few months [9,98] . Nevertheless, these “seronegative” individuals had strong CD4+ proliferative responses against LASV NP and GP antigens [60,61]. Importantly, these individuals cannot be protected against re-infection, but they are fully protected against disease indicating that a single primary infection with LASV confers long-term protection against LF [9].
The clearance of LASV does not correlate with antibody induction and LASV is a poorly neutralized virus. Clinical and experimental studies in infected NHPs showed that viremia and circulated antibodies were detected simultaneously in LF patients and infected animals (rev. [1,2,9]). At the time of hospital admission, LASV antibodies against NP or GP were not associated with survival or positive prognosis. Indeed, the presence of LASV antibodies detectable in IFA early in the course of the disease correlated with fatal outcome, not survival [99]. Recent studies confirmed the previous observation and showed that patients simultaneously containing LASV antigen and specific IgM antibodies have significantly higher chances (>4 times) to die in comparison with patients with IgM alone [100]. Taken together these results clearly indicate that protection and recovery are not associated with LASV antibody responses.
4. Non-Replication Competent Vaccine Platforms
4.1. “Killed” Vaccines and Virus-Like Particles
A favorable safety profile is the most attractive feature of inactivated (‘killed’) vaccines or virus-like particles, but these approaches in general have low immunogenicity and efficacy. To achieve desirable levels of protection these vaccines therefore require multiple prime-boost injections. Although this would be difficult to conduct in rural areas with poor healthcare infrastructures, such as the areas of West Africa where LASV is endemic, our group and others have tested the efficacy of this approach. In our experiments, extensive prime followed by multiple boost i.p. immunizations of CBA/Calac mice with highly purified, gamma-irradiated LASV preps resulted in some levels of protection against i.c. challenge, but this appeared to be mediated mostly by CMI (Godneva & Lukashevich, unpublished). However, although a similar approach in NHP induced excellent antibody responses, to NP and GP antigens as mentioned above, all vaccinated animals died after LASV challenge; postmortem titration did not reveal differences in tissue viral burden between vaccinated and non-vaccinated [97]. There is only one report published two decades ago in the Russian journal “Vopr. Virusol” [101] that describes protection of Papio hamadryads immunized with inactivated LASV preps against a subsequent LASV challenge. The authors demonstrated some level of protection against a relatively low challenge dose, 0.4 PFU. These results have not been independently reproduced or confirmed by other groups, and a model of human LF in hamadryads has not been developed.
Recent success of VLP-based vaccines against HPV motivated some researches to also apply this approach to LASV; they produced VLP by a transient expression of LASV GP, NP, and Z genes in mammalian cells [102]. Although some LASV VLP were immunogenic in mice after one prime and two boost immunizations, the VLP preps were contaminated with glycosylated proteins (30 to 220 kDa), presumably acquired during budding from the cell membrane or the Golgi apparatus [102]. Contamination concerns raise questions regarding a feasibility of this technology in manufacturing environment.
It is likely that the absence of viral RNA species in VLP contribute to the low effectiveness of LASV VLP. Specifically, successful viral vaccines (e.g; influenza vaccines) most often contain viral RNA that act as 'built-in' adjuvants. Specific receptors, RIG-I-like, Toll-like, and nucleotide-binding oligomerization domain-like receptors can detect specific nucleic acid patterns at early stages of antiviral responses, engage CMI, and thereby significantly enhance the adaptive immune responses [103,104,105,106,107]. Recent studies showed that RNA isolated from LASV was able to activate the IFN-β promoter [108]. The RNA-inducible TLR7-mediated signaling was required for activation of plasmocytoid DCs at early stages of infection to mount an effective adaptive immune responses that prevented development of persistent arenavirus infection [109,110].
Taken together, although “killed” virus or VLP can induce antibody responses in experimental subjects, the prospect of these approaches translating into an effect vaccine strategy is low. In spite of the fact that the current prospect of developing a killed LASV vaccine is low, novel adjuvant systems (e.g; ISCOMATRIX) [111] which significantly increase both antibody and CMI responses, can provide a new opportunity for a killed vaccine approach.
4.2. Peptide epitope-based vaccine approaches
Another potential approach is epitope-based vaccines. Using computer-assisted algorithms, five HLA-A2.1-binding LASV GP peptides and two LASV NP peptides have been identified [112,113]. When HLA-A*0201 transgenic mice were immunized with either LASV peptides GPC(42-50) or GPC(60-68), they were indeed protected against a subsequent challenge with a recombinant vaccinia virus that expressed LASV GPC [113]. In the recent study [114], a panel of HLA-A*0201-restricted peptides derived from the same region of the LASV GPC(441-449), LCMV GPC(447-455), JUNV GPC(429-437), MACV GPC(444-452), GTOV GPC(427-435), and WWAV GPC(428-436) was identified that displayed high-affinity binding to HLA-A*0201; and these peptides were cross-reactively recognized by CD8+T cells following LCMV infection or peptide immunization in HLA-A*0201 transgenic mice. Immunization of HLA-A*0201 mice with LASV GPC(441-449) or LCMV GPC(447-455) induced high-avidity CD8+- T-cell responses that were able to kill syngeneic target cells pulsed with either LASV GPC(441-449) or LCMV GPC(447-455) in vivo and provided significant protection against viral challenge with LCMV [114].
Although the peptide-based approach looks promising, especially for high-risk infectious agents, peptide vaccination is unlikely to be applicable for LASV because of significant safety concerns. As has been discussed, CMI responses, in particularly CD8+ T cells, play the major role to control primary infection; the CD8+ memory cell progeny also play a major role in protecting against subsequent re-infection. In addition to directly destroying infected cells, CTL (cytotoxic T lymphocytes) also release potentially cytotoxic cytokines (e.g; IFN-γ and TNF-α) that result in some degree of immunopathology. In fact, some of the systemic symptoms of viral infections are caused immune responses rather than by the infection itself. Although the precursor frequency of naïve T cells specific to any give epitope is low ~ 1:105 (102 cell/mouse spleen), acute infection can induce this frequency as high as107, which encompasses almost 50% of all CD8+ T cells in LCMV-infected mice, or 44% of all CD8+ cells in human PBMC after EBV infection. In non-naïve recipients, activation of these cells with an epitope-based peptide can induce immunopathology with serious consequences.
LCMV-specific peptide epitopes injected into LCMV-immune recipient mice activated pre-existing virus-specific CD8+ T cells; the resultant TNF-dependent immunopathology killed the vaccinated mice within hours [115]. Injection of vaccinia virus-specific peptide B8R20-27 after vaccinia virus infection resulted in very rapid hypothermia, although all mice recovered. This concern is highly relevant for LASV infection in West Africa where most LASV infections are asymptomatic with seroprevalence as high as 25%-55% [36,67,98]. Furthermore, clinically healthy LASV-exposed individuals who may have lost their antibodies still had robust CMI responses to LASV recombinant proteins [60,61]. Based on epidemiological observations in West Africa, it is very likely that these individuals have LASV-specific have memory CD8+ T cells to effectively control LASV after the re-infection. Administration of an epitope-based vaccine to these individuals can strongly re-activate the pre-existing CD8+ T clone and induce TNF-dependent immunopathology with serious clinical consequences [115]. In summary, while injection of a peptide vaccine into naïve individuals might probably be safe, peptide- based vaccination may have severe pathological consequences in individuals recently infected with the virus or in immune individuals previously exposed (perhaps unknowingly/asymptomatically) to the pathogen. In addition, (i) currently there is no licensed peptide vaccine, the concept is unproven; (ii) since in West Africa the variability of the human HLA genes is very high, a large number of peptides would potentially have to be included in the vaccine, raising a concern regarding the feasibility of this approach.
4.3. Alphavirus Vector-Based Vaccines
Alphaviruses are small positive-sense RNA viruses with genome of ~ 11.5 kb encoding two open reading frames for 4 non-structural proteins (nsp1-4, the replicase cassette) and structural proteins (capsid, c; and glycoproteins, gp). The nsp1-4 proteins are translated from the genomic RNA and structural proteins are translated from the subgenomic 26S mRNA which are produced in 10-fold molar excess [116]. Alphavirus replicon particles, RP, are generated by deletion of structural genes from the replicon RNA and replacing them with heterologous gene(s) of interest. Co-transfection of cells with replicon RNA and helper genes provided in trans results in the packaging of the replicon RNA into alphavirus-like particles. The most developed alphavirus replicon vectors are based on Venezuelan equine encephalitis virus (VEEV), Sindbis (SINV), and Semliki Forest virus (SFV) genetic backbones (rev. [117]).
To some extent, alphavirus replicon technology provides a reasonable compromise, in terms of safety and immunogenicity, between “killed” vaccines and replication-competent platforms. Alphavirus RP are single-cycle, replication-defective vehicles (vectors). They are not able to spread beyond the initially infected cells, but can deliver and transduce the gene(s) of interest in target cells. Direct comparison of the immune responses induced by alphavirus-vectored vaccines and inactivated vaccines in mice showed clear advantage of aphavirus-vectored platform [118]. The alphavirus RP also offers attractive features in terms of safety, immunogenicity, and efficacy and currently numerous vaccine candidates are in pre-clinical and clinical development (rev. [117]).
One of the features of alphavirus RP is tropism to DCs. In contrast to “nucleic acid-free” VLP that have limited capacity to induce innate immunity, alphavirus RP induce robust innate immune responses. In recent study VEEV-based RP alone (without a transgene, “null” RP) were used as an adjuvant and dramatically improved the immunogenicity and protection efficacy of a licensed inactivated influenza vaccine in rhesus macaques [119]. Notably, the “null” RP induced mucosal responses even when the particles and immunogen were delivered via systemic routes [120,121]. It was also demonstrated that the alphavirus RP acted as a potent adjuvant to promote CD8+ T cells responses to co-delivered antigen [122], the feature which is especially attractive for LASV vaccine design.
VEEV-based replicon technology was successfully used to make mono- and bivalent vaccines expressing LASV NP, GPC and Ebola virus (EBOV) GP genes [96,123]. LASV genes were cloned downstream from the VEEV replicase and the 26S promoter in the transcription plasmid that contained the VEEV replicon cDNA. The LASV-replicons and two helper RNAs (c and gp) were prepared by in vitro transcription of the recombinant plasmids using T7 RNA polymerase and were used for co-transfection of BHK-21 cells. At 30 h post-transfection, the titers of the LASV-NP and LASV-GPC replicons in the medium of cotransfected cells were 107 and 108 IU/ml, respectively. The LASV RPs were negative for VEEV and expressed LASV NP and GPC. All strain 13 guinea pigs immunized with LASV-NP, LASV-GPC, LASV-NP plus LASV-GPC (5 animals/group) were protected against challenge with 160 LD50 of LASV (Jos). Low (at the level of detection), transient viremia was detected at day 7 after challenge in 3 animals vaccinated with LASV-GPC, and in one guinea pig in the LASV-NP group and in the LASV-GPC plus LASV-NP group [96]. Notably, immunization with RP expressing LASV NP and GPC genes induced robust antibody responses detected by IgG ELISA but not in plaque neutralization assay.
The VEEV-based replicon technology was also used to make a bivalent replicon for simultaneous expression of glycoprotein genes of LASV and EBOV. Vaccination of guinea pigs with dual-expression particles protected the animals against challenges with both viruses, LASV an EBOV [96].
The results summarized above indicate that replication-incompetent vectors that target antigen-presenting cells to deliver rationally-designed multivalent cross-protective arenaviral immunogens represent a feasible and attractive strategy. In the 1st generation of alphavirus-based replicon technology [123,124,125], replication-competent virus (RCV) can be generated by recombination of helper RNAs with replicon RNA (Figure 2). During packaging, recombination could potentially occur between replicating vector and helpers by template-switching mechanism, which could lead to regeneration of RCV, a live VEEV virus. Although helpers were split into c and gp parts (Figure 2) and attenuating mutations were introduced into helpers, the 1st generation of VEEV vectors required a BSL-3 containment for vaccine manufacturing [123].
To minimize the possibility of RCV regeneration and improve vaccine safety, a backbone of human TC-83 vaccine (IND No. 142) was used for making replication expression vectors and the packaging helper system (Figure 2) in the 2nd generation of this technology. The alphavirus 26S promoter for helper genes has been substituted with alphavirus-non-related CMV promoter and packaging helpers have been replaced with non-replicating constructs. These modifications will eliminate the possibility of
![Viruses 04 02514 g002]() RCV generation. ). Additionally, the replicon expression vector has been re-designed to clone up to 5-kb-foreign insert and to express two (or more) foreign antigens. The multivalent feature of this system is certainly beneficial to optimize LASV vaccine formulation (e.g; simultaneous expression of GPC and NP genes); to address LASV genetic diversity (e.g; to express GPC from distantly related clades, I and IV); and to enhance immunogenicity of experimental vaccines (e.g; to express “wild-type” GPC for conventional antigen presentation and metabolic stable GPC for cross-priming CD8+ T cell responses). This technology is currently used to manufacture a bivalent cross-protective LASV vaccine for preclinical studies.
RCV generation. ). Additionally, the replicon expression vector has been re-designed to clone up to 5-kb-foreign insert and to express two (or more) foreign antigens. The multivalent feature of this system is certainly beneficial to optimize LASV vaccine formulation (e.g; simultaneous expression of GPC and NP genes); to address LASV genetic diversity (e.g; to express GPC from distantly related clades, I and IV); and to enhance immunogenicity of experimental vaccines (e.g; to express “wild-type” GPC for conventional antigen presentation and metabolic stable GPC for cross-priming CD8+ T cell responses). This technology is currently used to manufacture a bivalent cross-protective LASV vaccine for preclinical studies.
Figure 2.
Advanced alphavirus-based vector technology. The 1st generation was based on Venezuelan equine encephalitis virus (VEEV) and required BSL-3 containment. Replication-competent virus (RCV) can be generated during a recombination event. In the 2nd generation, vectors use human vaccine VEEV TC-83 vaccine backbone and transcription of helper genes is controlled by non-related CMV (cytomegalovirus) promoter. Helper proteins (capsid and glycoproteins, E1&E2) can be made from two mono-cistronic CMV-plasmids or from one CMV-bicistronic plasmid (see text). Recombination events are minimized and BSL2 containment is required for production ([126]; Pushko & Lukashevich, unpublished.
Figure 2.
Advanced alphavirus-based vector technology. The 1st generation was based on Venezuelan equine encephalitis virus (VEEV) and required BSL-3 containment. Replication-competent virus (RCV) can be generated during a recombination event. In the 2nd generation, vectors use human vaccine VEEV TC-83 vaccine backbone and transcription of helper genes is controlled by non-related CMV (cytomegalovirus) promoter. Helper proteins (capsid and glycoproteins, E1&E2) can be made from two mono-cistronic CMV-plasmids or from one CMV-bicistronic plasmid (see text). Recombination events are minimized and BSL2 containment is required for production ([126]; Pushko & Lukashevich, unpublished.
4.4. DNA Immunization
The authors of the first publication on LASV DNA vaccine [127] concluded that “DNA vaccine against LASV should encode as much of the proteome as possible, including both Lassa NP and GP”. In recent review, Grant-Klein et al. [128] noted that LASV DNA expressing GPC fully protected guinea pigs and cynomolgus macaques against LASV challenge. However, no results or details were provided. A LASV DNA vaccination approach was tested in LCMV murine challenge model based on the existence of common CD8+ T cell epitopes inducing cross-protective immunity. This model provides a quick and reliable test of a vaccine’s ability to induce CMI. It was shown that LCMV NP expresses the immunodominant CTL epitope (NP118-126) for H-2d mice [129], and within the epitope, a minimal tetrapeptide GVYM is identical for LCMV, LASV, MOPV and plays an important role in CTL recognition [130]. Recombinant LASV NP protein expressed in vaccinia or in Salmonella protected 33% of mice from LCMV challenge, and the protection was associated with cross-reactive CTL and proliferative responses [131,132]. Similarly, a DNA vaccine expressing LASV NP showed partial (50%) cross-protection against LCMV and Pichinde virus (PICV) challenges in mice. DNA vaccination with a single epitope (LASV NP118-126) also partially protected mice from LCMV but not from PICV i.c. challenge [127]. Protection induced by the LASV NP118-126 minigen vaccine in this challenge model provided some rational for making multi-CD8+ CTL epitope constructs (“string of beads” vaccines [133]). However, these approaches share the negative consequences of the application of an epitope-based strategy in population with high prevalence of individuals previously exposed to LASV (see above) and therefore may not be applicable for LF vaccine.
Electroporation has recently emerged as a powerful tool to improve immunogenicity of DNA vaccines. In animal studies, electroporation enhanced efficacy of DNA vaccines by 10-1000-fold. However, phase I/II clinical trials showed the most common adverse effects are pain and bleeding at the injection sites. Electroporation is likely to be unsuitable for prophylactic vaccination, especially in children. However, this approach can be applicable for therapeutic cancer vaccines [134].
4.5. Rationally Designed Replication-Competent Vaccines
Replication-competent, “live-attenuated”, vaccines are among the most cost-effective and widely used public health interventions. The vaccines for smallpox, polio, and yellow fever dramatically reduced the incidence of these infectious diseases. However, successes in life science, biotechnology, and public health changed the emphasis on the effectiveness of vaccines to the safety of vaccines. This paradigm shift has led to the irony that the present regulatory standards would probably prevent the licensure of live-attenuated vaccines today. Currently, advances in molecular virology and rational design provide new opportunities for development of the next generation of these vaccines that optionally balance safety and effectiveness [135].
In most cases, LASV infection does not result in manifested disease, with only about 20% of infected people displaying the symptoms of the disease, which range from flu-like symptoms to fatal LF [2,9]. Long-term epidemiological observations in West Africa suggest that in immunocompetent adults, a single naturally “attenuated” infection can induce lengthy protection against fatal LF, and re-infection with heterologous strains will provide boosting effects [36,136]. While a high level of annual re-infection (up to 18%) is one of the characteristic features of LASV infection [5,98,136], a second clinical attack has never been documented [9].
There are several reasons to justify a replication-competent, “live” vaccine as an attractive approach to control LF: (i) CMI plays the major role in LF patient recovery and protection; (ii) a live vaccine provides the most effective natural pathway to process and present protective antigens to MHC molecules; (iii) epidemiological observations in LF endemic areas of West Africa provide evidence that a single (survived) exposure will induce long-term protection against disease [36]; (iv) a vaccine candidate formulated to contain both LASV NP and GP antigens will induce a broad cross-reactivity and a large pool of CD4+ memory T cells against all phylogenetic groups of LASV [60,61,137]; (v) a single-shot immunization approach is a crucial for population of remote rural areas of West Africa, which has a very limited medical infrastructure and where implementation of prime-boosting immunization is not practical.
The first vaccine for the prevention of viral hemorrhagic fevers caused by arenaviruses is Candid #1, a live attenuated vaccine. This vaccine is safe, highly immunogenic and efficacious. In 2007, this vaccine was included into the Argentine National Immunization Plan and significantly reduced morbidity and mortality caused by JUNV infection [138]. Currently there are four replication-competent LASV vaccine candidates based on vaccinia virus [94,139,140], vesicular stomatitis virus (VSV) [141,142], MOPV [48,137,143], and yellow fever 17D [144,145] vectors. All these vaccine candidates have been tested in NHP and will be described in more detail.
4.5.1 Recombinant vaccinia virus expressing LASV antigens
The first experimental vaccine successfully tested in animals was a recombinant vaccinia virus (Lister) expressing NP of LASV strain GA391 isolated from a patient in northern Nigeria [94]. The NP gene was placed in the transfer vector pGS20 and subsequently inserted into the thymidine kinase gene of vaccinia under control of the 7.5K promoter. Six outbred (Dunkin-Hartley) guinea pigs inoculated with of 107.5 PFU (s.c.) of recombinant vaccinia were fully protected against fatal challenge with 104 TCID50 (tissue culture infectious doses) of LASV GA391. Protected animals had no clinical signs of the disease and viremia was not detected.
Cloning of both LASV (Josiah) major antigens, NP and GPC, into the vaccinia vector (NYBH) gave similar results as described above; 94% of outbred guinea pigs vaccinated with LASV-NP (V-LSN) and 79% vaccinated with LASV-GPC (V-LSG) were protected against LASV (Jos) challenge [146]. Protective efficacy of vaccinia recombinant V-LSG was confirmed in rhesus monkeys (Macaca mulatta) [147]. Four animals were vaccinated with 109 PFU of V-LSG and two rhesus monkeys received MOPV (104 PFU, a positive vaccination control). Vaccinated animals were challenged on day 37 with LASV (Jos, 104 PFU), and survived the challenge. Low transient viremia (102-103 PFU/ml) was detected in all survived animals on day 7-9 after challenge. In spite of complete recovery, V-LSG-vaccinated animals developed a mild fever, some chemical and hematological abnormalities (depressed platelet function, lymphopenia and neutrophilia) which were not observed in MOPV-vaccinated/LASV-challenged animals. After vaccination, only two V-LSG animals developed antibodies to LASV GP, and no serum from any animals after vaccination had any neutralizing activity, which again emphasizes the importance of CMI over humoral immunity.
A broader study in 44 NHPs (28 rhesus and 16 cynomolgous) that summarized vaccination/challenge experiments with recombinant vaccinia that expressed different parts of GPC, NP, provided more detail on the efficacy of vaccinia-based recombinant vaccines and contribution of individual antigens to protection [140]. These results showed that both glycoproteins, GP1 and GP2, are required for protection, and that the full length GPC is necessary and sufficient to protect animals against challenge.
In contrast to results in guinea pigs, 12 out of 15 animals vaccinated with NP died (20% survival), although this vaccination developed a strong antibody response before challenge. Animals vaccinated with GPC had low antibody titers before challenge, but 17 from 19 vaccinated were fully protected (88%). The highest level of protection (90%) was observed when animals received all proteins encoded by the LASV S RNA (V-LSG/N or V-LSG + V-LSN). In these two groups, the lowest levels of transient viremia, comparable with levels in MOPV-vaccinated group (a vaccination control), were detected. No neutralizing antibodies were detected in any animal groups before or after challenge. This study confirmed that antibody plays a minor, if any, role in virus clearance and protection. It was also demonstrated that challenge dose, as well as the interval between vaccination and challenge can affect outcome of the experiments [9,36,140]. This study also highlighted a controversial role of LASV NP in protection and the relative importance of a reliable and validated animal model for evaluation of LASV immunogenicity and efficacy (see below).
4.5.2. Replication-competent VSV vectors expressing LASV glycoproteins
Initially, recombinant vaccinia virus expressing LASV genes provided promising results and helpful information for LASV vaccine development. However, this vaccine platform is no longer considered as acceptable because of potential safety issues associated with the immunosuppressive phenotype of the vector. The prevalence of immune-suppressed individuals is especially high in Sub-Sahara Africa. Nigeria has the highest number of clinical cases of LF and it also ranks as the second worst HIV-1-affected country in the world (after South Africa). In our preliminary studies, we found that LASV sero-prevalence was 3-times higher in HIV-positive individuals than in the HIV-negative group (Abimiku & Lukashevich, unpublished) indicating that both infections co-exist in Nigeria and can target the same population.
Attenuated recombinant VSV (rVSV) vector was proposed as a reasonable alternative to a vaccinia virus platform. The VSV is a negative-stranded, non-segmented RNA virus belongs to the Rabdoviridae, genus Vesiculovirus. The virus infects a wide range of animals, replicates very well in different types of cells, and induces a strong cytopathic effect. The genomic RNA encodes five major proteins, glycoprotein (G), matrix protein (M), nucleoprotein (N), large protein (L) and phosphoprotein (P). There is a powerful molecular biology tool to genetically manipulate VSV; the rVSV lacking its own G gene has been widely used to produce pseudotypes with envelope proteins of heterogeneous viruses (rev. [148]). The G is the major pathogenic factor and replacement of VSV G with a heterogeneous type I transmembrane glycoprotein results in generation of attenuated rVSV, incorporating the foreign glycoprotein on the bullet-like VSV surface. Based on this feature and the ability to tolerate foreign transcription units, rVSV was successfully used as a promising vaccine vector for varieties of pathogens; among them, rVSV expressing EBOV and MARV glycoproteins, were extensively tested in NHPs with encouraging results (rev. [149,150]).
The filovirus GP cloning strategy applied for LASV GPC resulted in generation of recombinant VSV∆G/LASVGP particles with bullet-shaped nucleocapsid morphology [141]. For both recombinant viruses, (VSV∆G/LASVGP and VSV∆G/ZEBOVGP), growth kinetics in Vero cells at high m.o.i. were attenuated, reaching maximum titers 24 and 36 h post-infection, respectively. Western blot analysis detected the LASV glycoprotein precursor, GPC, in infected cells and proper cleavage into GP1 (44 kDa) and GP2 (36 kDa). Immunoelectron microscopy using monoclonal antibodies against LASV GP1 and GP2 confirmed expression of LASV glycoproteins on surface of recombinant virus particles [151]. As expected, replacement of VSV gene with LASV GP resulted in cell tropism changes. The wild-type VSV infects and replicates in human T-cell leukemia cells (Jurkat), but these cells are not susceptible for LASV and EBOV. Recombinant VSV∆G/LASVGP and VSV∆G/ZEBOVGP failed to replicate in these cells as well [141].
Recombinant VSV∆G/LASVGP (2 x 107 PFU) was inoculated (i.m.) into four cynomolgus macaques [151]. Immunized animals did not display any clinical symptoms. Twenty-eight days after immunization, vaccinated and control monkeys were challenged with 104 PFU of LASV (Jos). The control animals developed a severe LF and were euthanized on days 11 and 12. All vaccinated animals were fully protected and survived the LASV challenge without clinical manifestations. Hematology and blood chemistry analysis revealed slight perturbations in platelet numbers and in plasma ALT levels in vaccinated animals after LASV challenge. Although viremia was not detectable after immunization, LASV was transiently detected in all vaccinated animals at low (102 PFU/ml) to moderate (104 PFU/ml) levels, indicating that immunization did not induce sterilizing immunity. All 4 vaccinated macaques developed moderate- to high levels of IgG ELISA antibodies and low levels of neutralizing antibodies before challenge. CMI responses were detected by intracellular staining of CD4+ and CD8+ lymphocytes for secretion of IFN-γ and TNF-α. Secretion of these cytokines was found in CD8+ cells only in one vaccinated monkey before challenge. LASV challenge markedly increased antibody responses (ELISA, neutralization) in all animals. Surprisingly, LASV challenge stimulated CMI responses only in 3 of 4 protected animals, indicating that protection at least in one animal was not associated with activation of CD8+ or CD4+ T cells. Although mechanism of protection induced by rVSV∆G/LASVGP is not still clear and some safety concerns still need to be addressed, this vaccine candidate is well positioned to further development. Recent progress with filovirus vaccines based on the same technology should also provide a helpful guidance.
4.5.3. Reassortant vaccine platform, MOP/LAS (clone ML29)
The bi-segmented feature of the arenavirus RNA genome indicates that these viruses can produce reassortants during co-replication in vitro. Nevertheless, natural reassortance between arenaviruses has not been reported; although there is an evidence for natural genetic recombination within the S RNA segment of the NW arenaviruses [152]. Intertypic reassortants within LCMV [153,154,155] and PICV [154,156] species and between two different species (LASV and MOPV) [48,49], were generated in vitro; these reassortant viruses were successfully used for coding assignment and for understanding the molecular basis of virus pathogenicity. Reassortant analysis indicates that the L RNA is important for high levels of virus replication in vivo and it is associated with acute disease in experimental animals [155,156].
The MOP/LAS reassortant, clone ML29 [12,157], was designed to keep the attenuated profile of MOPV, but to induce a robust cross-protective CMI responses against genetically-distant LASV isolates. Clone ML29 was selected from a library of MOPV/LASV reassortants and encodes the major antigens (NP and GPC) of LASV, as well as the L protein (RdRp) and Z protein of MOPV [48]. Since clone ML29 has 70% of MOPV genome, it has the same risk classification as the vector virus, MOPV. MOPV is considered risk group 2 by the EU biosafety regulation and risk group 3 by the CDC.
While co-infection of Vero cells with MOPV and LASV resulted in production of stable reassortants, our attempts to isolate viable reassortants after co-infection of cells with LASV and LCMV failed. Recent experiments using minireplicon systems developed for these viruses explained these results. The minigenoms (MGs), NPs, and L proteins of LASV and MOPV were exchanged and interacted homologously and heterologously without loss of function. L proteins of LASV and MOPV were also active in interaction with LCMV NP. However, the LCMV L protein required only homologous NP for its activity [47]. Transcriptional replicon activity of MGs was measured via expression of the Ren-Luc reporter. When activity of 3 minireplicons was compared, the 10-fold-reduced activity of the MOPV system compared to the LASV and LCMV systems was found [47]. Interestingly, the substitution of the MOPV 3′-UTR by that of LASV increased the expression of Ren-Luc from the MG suggesting that translation is initiated less efficiently at the MOPV 3′-UTR than at the LASV 3′-UTR. It is intriguing to find evidence how this “attenuated” initiation translation is associated with attenuated phenotype of MOPV.
Eighteen mutations distinguish the ML29 genome from the parental arenaviruses, MOPV (L RNA) and LASV (S RNA) and likely additionally contributed to the attenuated phenotype [137,157] (Table 1). The ML29 genome was stable during 12 passages in tissue cultures and all these mutations were preserved as it was shown by deep sequencing of ML29 isolates. Among 18 mutations, noncoding transversions A6C, U8G and A7264C, U7266G were located within the 3’ and 5’ termini of the L RNA (derived from MOPV) and one mutation, G3328A was identified within the long 3’ non-coding region of the S RNA (derived from LASV) of ML29. A recently developed LASV reverse genetic system was applied to investigate the contributions of noncoding regions of the LASV S RNA on growth kinetics in vitro and virulence in mice [158]. This approach showed that deletions of large portions of these noncoding sequences resulted in attenuated LASV replication in vitro and in vivo.
The terminal 19 nucleotides of each RNA segment are highly conservative among arenaviruses and represent the promoter directing the virus genes expression and genome replication [159,160]. These sequences are mostly complementary to each other, since perfect complementarity within the terminal UTRs is required for efficient RNA replication [161]. Mutational analysis of LASV promoter in the S RNA MG system showed that this promoter coordinates transcription and replication. It contains two regions, a sequence-specific region (residues in positions 1-12) and a variable complementarity region (positions 13-19) [159]. Mutations distinguishing the parental MOPV L RNA sequence from the analogous terminal sequences of the ML29 L RNA were located at positions 6 and 8. Mutations in these positions were tolerated, consistent with some natural variability among arenaviruses. Notably, these mutations suggest that the predicted pan-handle secondary structure of the ML29 L promoter is more stable than those in the parental viruses, LASV and MOPV [12,157].
Mutations in the coding region of the L RNA resulted in 3 non-conservative amino acid substitutions at positions 851(Tyr→Asn), 1233 (Arg→Gly), and 2136 (Asp→Asn). Only the R1233G
mutation was located within the putative RdRp module and mapped between motif A, Asp-X(2)-Lys-Trp, and motif B, Gly-X(5)-Ser. These motifs represent stretches of highly conserved sequences that are linked by less conserved variable regions [162]. The Y851N substitution was located between N-terminal and an RdRp domain located in the central part of the molecule. A large scale mutagenesis approach found that an N-terminal region of LASV L protein plays a crucial role in transcription but not in replication of the LASV genome. The so called “L1 domain” (from position 1 to 300) can be a part of an endonuclease involved in putative a cap-snatching mechanism required for mRNA synthesis [163]. The N-terminal domain of MOPV L protein seems to be involved in the similar activity. The functional role of a C-terminal region where a D2136N substitution was found is currently unknown.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations/Substitutions | Location | Putative function | Possible effects (References) |
|---|---|---|---|
| A6C, U8C | L RNA, 5’, 3’ NCR | Form panhandle | Enhance promoter stability [12] |
| A7264C | |||
| A7264C | |||
| G3328A | S RNA, 3’ NCR | Unknown | Deletions in the 3’NCR attenuate replication [158] |
| Y851N | L protein, N-term. | Cap-snatching | Attenuated transcription/ replication [162,163]? |
| N173S | NP, central domain | Interaction with RNA | Unknown |
| A485D | NP, C-terminal | Exonuclease | IFN modulation [164,165] |
| K272E | GP2, N-terminal | Fusion | Affect fusion/post-fusion events? |
The S RNA of ML29 was derived from LASV and differs in sequence by six mutations. These mutations resulted in three amino acid substitutions, two of them were located in NP and the 3rd mutation was identified in N-terminal domain of GP2. The homologous NP mutation, N173S, was mapped in the central region of the protein, and a non-conservative substitution, A485D was located in the C-terminal region of NP which was critical for immunosuppressive function [166,167,168]. Crystal structure of LASV NP showed that this mutation is located between two aspartic acid residues at positions 466 and D533 which involve in formation of active site of the putative LASV NP exonuclease associated with the NP ability to suppress translocation of IRF-3 and to block IFN activation [164,165,169]. This site contains two critical residues, D389 and G392, and recombinant LASV mutants carrying mutations at these residues were severely altered in their ability to suppress IFN in DCs and macrophages.
Both viruses, LASV and ML29, have identical surface-exposed GP1 responsible for interaction with cell receptors; while ML29 GP2 is different from LASV GP2 by the K272E substitution located between two fusion domains. The transmembrane GP2 is the class I fusion protein. We for the first time demonstrated the fusion activity of an artificial peptide derived from highly conserved hydrophobic region at amino acid positions 276-298 [170]. Later, Klewitz et al. [171] confirmed these results and found that the N-terminal stretch, residues 260-266, located downstream of the GPC cleavage site is also involved in the fusion activity. Notably, all LASV strains analyzed so far have mutations between two domains with fusion activity, at positions 272-274 (K-D-T for LASV Josiah GP2). In ML29 GP2 Lys to Glu substitution introduces two negatively charged groups in positions 272-273 (Asp-Glu) distinguishing the MOP/LAS reassortant from other LASV strains analyzed so far [137,172]. These changes significantly affect the predicted GP2 structure (Figure 3) by refolding and exposure of C-terminal part of the GP2 with transmembrane domain and a cytoplasmic tail. This rearrangement of GP2 transmembrane domain can potentially affect fusion and/or post-fusion events during ML29 entry. Interestingly, the F427I substitution found in GP2 transmembrane domain
![Viruses 04 02514 g003]() of JUNV Candid #1 vaccine seems to be involved in the destabilization of the GP metastable conformation and probably contributes to attenuation [174]. At least virus entry/fusion directed by Candid #1 GP2 was significantly less affected by NH4Cl (to block the endosomal acidification) than the entry mediated by wild type GP2 [174]. Key role of the F427I substitution in attenuation of Candid #1 was recently confirmed by reverse genetics [175] and this mutation was proposed to be a molecular signature of JUNV attenuation [174]. Interestingly, we [12] and others [176] also observed different effects of ammonium chloride treatment on LASV and MOPV replication kinetics in cell cultures.
of JUNV Candid #1 vaccine seems to be involved in the destabilization of the GP metastable conformation and probably contributes to attenuation [174]. At least virus entry/fusion directed by Candid #1 GP2 was significantly less affected by NH4Cl (to block the endosomal acidification) than the entry mediated by wild type GP2 [174]. Key role of the F427I substitution in attenuation of Candid #1 was recently confirmed by reverse genetics [175] and this mutation was proposed to be a molecular signature of JUNV attenuation [174]. Interestingly, we [12] and others [176] also observed different effects of ammonium chloride treatment on LASV and MOPV replication kinetics in cell cultures.
Figure 3.
The predicted structure of GP2 glycoprotein of ML29. The non-conservative K272E substitution (marked by star) located between two fusion domains at N-terminus introduced two negatively charged groups in positions 272-273 (Asp-Glu) resulted in refolding and exposure of transmembrane and cytoplasmic domains. Protein structure prediction was performed using the Phyre server (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) [173].
Figure 3.
The predicted structure of GP2 glycoprotein of ML29. The non-conservative K272E substitution (marked by star) located between two fusion domains at N-terminus introduced two negatively charged groups in positions 272-273 (Asp-Glu) resulted in refolding and exposure of transmembrane and cytoplasmic domains. Protein structure prediction was performed using the Phyre server (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) [173].
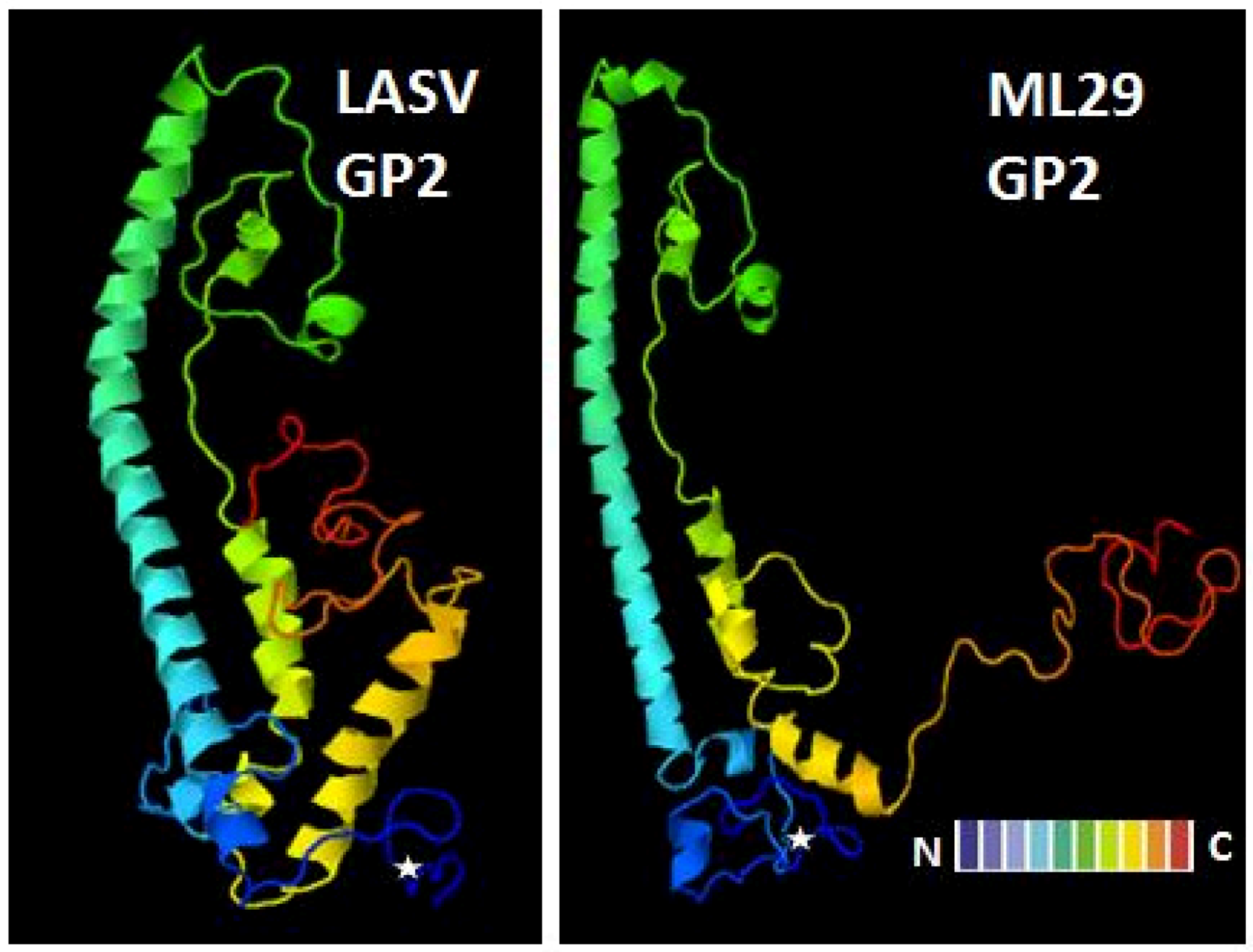
The immunogenicity and efficacy of ML29 vaccine candidate was tested in all available animal models to comply with the FDA Animal Rule [177] and included mice, guinea pigs, and NHP (rhesus macaques and common marmosets). Although mice do not accurately model human LF disease, they can provide an economical assay for vaccine potency in terms of capacity to elicit CMI and recentstudies demonstrated correlation between LASV-specific CD8+ cytotoxicity in vivo and ML29-induced protection [178]. The ML29 immunization with 150 PFU and higher produced immune splenocytes that completely protected animals against a fatal LASV challenge [137]. Protective T lymphocytes were detected in spleen as early as 2 days after vaccination. The cells collected on day 5 and 7 protected 60 and 100% of mice, respectively. Protection was dose-dependent and required 3 x 107splenocytes to protect all recipient mice against fatal LASV challenge. LASV infection of CBA mice resulted in rapid accumulation of the virus in tissues and the death of animals at day 6-7 after i.c. inoculation [179]. Injection of the ML29-immune splenocytes effectively eliminated the virus from tissues of challenged animals. At day 3 after immune lymphocyte transfer, the virus was undetectable in spleen and other tested tissues (brain, liver, kidneys) of recipient mice. The effective virus clearance seems to be the main mechanism of protection of mice treated with ML29-specific T lymphocytes.
A single injection of ML29 provided full protection of strain 13 guinea pigs against more than 3,300 LD50, animals did no express any clinical or biochemical abnormalities during the observation period (70 days). Meanwhile, in MOPV-vaccinated animals, a transient elevation of AST and alkaline phosphatase in plasma was observed at week 3 after challenge, suggesting liver injury [137]. Histology studies did not reveal lesions in tissues of ML29-vaccinated animals that were challenged with LASV.
Genetic diversity of LASV has been a great challenge for vaccine development. The highest sequence differences (based on partial NP sequences) were found between lineages II (803213/NIG/74/H) and IV (JOS/SL/76/H), 11.0-14.4%. To determine whether the ML29 vaccine can protect strain 13 guinea pigs from distantly related LASV, vaccinated animals were challenged with LASV-JOS or with LASV-NIG. The S RNA of ML29 derived from LAS-JOS strain [137,157] and challenge with this virus was considered as a homologous vaccination-challenge protocol.
As seen in Table 2, all ML29-vaccinated animals were completely protected against challenge with two LASV strains, Josiah/SL (group 7) and 803213/NIG (group 8), and had no clinical manifestations of the disease. However, ML29-vaccinated animals were not protected from challenge with LCMV-WE, although vaccinated animals lived longer than animals in the control group, 16-21 days vs 13-14 days (group 9). These observations indicate that ML29 vaccination can induce some levels of cross-protection against LCMV-WE.
Table 2.
Results of vaccination and challenge experiments in guinea pigs [180].
| Animal Group | Challenged virus | Dose, PFU | Vacc/chal interval, days a | No. survived/No. infected | Survival, % | Day of death | |
|---|---|---|---|---|---|---|---|
| No vaccination | |||||||
| 1 | LASV-Jo | 10e+1 | na b | 0/4 | 0 | 15-17 | |
| 2 | LASV-Jo | 10e+3 | na | 0/5 | 0 | 15-16 | |
| 3 | LASV-803213 | 10e+3 | na | 0/5 | 0 | 13-15 | |
| 4 | LCMV-WE | 10e+3 | na | 0/5 | 0 | 13-14 | |
| The ML29 conventional vaccination (challenge on day 30) | |||||||
| 5 | 10e+2 | no challenge | na | na | 6/6 | 100 | na |
| 6 | 10e+6 | no challenge | na | na | 6/6 | 100 | na |
| 7 | 10e+3 | LASV-Jo | 10e+3 | 30 | 6/6 | 100 | na |
| 8 | 10e+3 | LASV-803213 | 10e+3 | 30 | 5/5 | 100 | na |
| 9 | 10e+3 | LCMV-WE | 10e+3 | 30 | 0/6 | 0 | 16-21 |
| The simultaneous vaccination/challenge experiments (challenge on day 0 and 2) | |||||||
| 10a. | 10e+6 | LASV-Jo | 10e+1 | 0 | 5/5 | 100 | na |
| 10b. | 10e+6 | LASV-Jo | 10e+1 | 2 | 3/5 | 60 | 10c,15 |
| 11a. | 10e+6 | LASV-Jo | 10e+3 | 0 | 4/4 | 100 | na |
| 11b. | 10e+6 | LASV-Jo | 10e+3 | 2 | 4/5 | 80 | 10 c |
| 12a. | 10e+2 | LASV-Jo | 10e+3 | 0 | 3/4 | 75 | 14 |
| 12b. | 10e+2 | LASV-Jo | 10e+3 | 2 | 3/4 | 75 | 16 |
| 13 | 10e+6 | LASV-803213 | 10e+3 | 0 | 3/5 | 60 | 12,17 |
| 14 | 10e+6 | LCMV-WE | 10e+3 | 0 | 0/5 | 0 | 14-16 |
a Animals were s. c. vaccinated with the ML29 reassortant (day 0) and challenged simultaneously on day 0, 2, 30 after vaccination. Death or survival past 21 days was set up as an endpoint. Amino acid difference between LASV-Jo and LASV-803213 is the highest within LASV genetic lineages I-IV.
b Non applicable; c Non-LASV-specific death (inappropriate anesthesia).
Several examples of natural reassortment between genetically-related bunyaviruses raised the concern that vaccination with an attenuated reassortant vaccine in endemic areas will affect the natural pattern of the disease and enhance pathogenicity. To address this concern challenge and vaccine application was performed simultaneously (day 0). All animals vaccinated with 1 x 106 PFU of ML29 survived after simultaneous challenge with low (group 10a) or high (group 11a) doses of LASV-Josiah. Importantly, the vaccination also protected 60% animals from challenge with high dose of the distantly-related strain, LASV-803213/NIG (group 13). Vaccination with low dose, 1 x 102 PFU of ML-29, protected 75% animals against challenge with high dose of LASV-Josiah. Vaccination two days before challenge was also effective protecting more than 60-80% of the guinea pigs against death.
In ML29-vaccinated animals challenged on day 30 with LASV-Jos or with distantly-related Nigerian strain viremia was not detectable by conventional plaque assay, co-cultivation on Vero cells, or by qRT-PCR at all tested time-points, on day 4, 8, 12, 21 after challenge. Therefore, this demonstrates that those animals challenged 30 days after vaccination were not only completely protected against disease but also against LASV infection suggesting that vaccination with ML29 induces sterilizing immunity. In partially protected animals clinical parameters were not as severe as in non-vaccinated animals and partial recovery on day 21 [180]. Non-neutralizing IgG ELISA antibodies against LASV, predominantly anti-NP, were detectable at the end of the 1st week of vaccination and peaked shortly after challenge. These antibodies apparently do play major role in the recovery and protection [180].
Protection of animals after simultaneous vaccination-challenge justified a post-exposure treatment of LASV-exposed animals. As seen in Figure 4, 80% guinea pigs were protected against fatal LF when animals were treated with ML29 two days (48 hours) after LASV challenge. This treatment resulted in clinical (temperature and weight) and biochemical (albumin, AST/ALT) improvements and recovery.The similar experiments are under way to test efficacy of ML29 post-exposure treatment of rhesus macaques fatally infected with LASV (R. Carrion, personal communication).
Figure 4.
(A), Survival rate of strain 13 guinea pigs (4 animals per group) fatally infected with LASV (Jos, 1000 PFU, s.c.) and treated on day 2 and 4 after challenge with ML29 (1000 PFU, s.c.); (B), ML29 preventive efficacy in common marmosets. Animals (6 animals per group) were vaccinated with ML29 and challenged on day 30 with LASV (Jos) [143].
Figure 4.
(A), Survival rate of strain 13 guinea pigs (4 animals per group) fatally infected with LASV (Jos, 1000 PFU, s.c.) and treated on day 2 and 4 after challenge with ML29 (1000 PFU, s.c.); (B), ML29 preventive efficacy in common marmosets. Animals (6 animals per group) were vaccinated with ML29 and challenged on day 30 with LASV (Jos) [143].
Mechanisms of protection in postexposure treatment are not clear. Currently at least two hypotheses are under evaluation: (i) the ML29 L RNA has a more stable promoter and higher affinity to RdRp and effectively outcompete the LASV during co-replication in target cells (DCs, macrophages); (ii) the ML29 treatment will rescue fatally infected animals by activation of ML29-specifi innate immune responses overcoming LASV infection.
In rhesus macaques, a single injection of 1,000 PFU (s.c.) of ML29 resulted in induction of primary virus-specific T cells capable of secreting IFN-γ in response to homologous and heterologous antigen stimulation [137]. These cells can be detectable in peripheral blood and in spleen on day 7 after immunization. On day 28 after vaccination, among the ML29-specific cells, up to 13% of T cells cross-reacted in response to closely related viruses, LCMV-WE or MOPV [137]. ML29 immunization resulted in antibody responses detected in ELISA at week 2 after immunization and with an endpoint dilution titer of 1:62,500 on day 28. Neutralization antibodies were undetectable (<1:20) at this time-point.
Table 3.
Viremia in marmosets vaccinated with ML29 and challenged on day 30 with LASV a [143].
| Animal No | Time, days after challenge: | |||||
|---|---|---|---|---|---|---|
| 0 | 5 | 15 | 17-19 | 20 | 35 | |
| Non-vaccinated | ||||||
| CJ25391 | <1.0 | 2.95 | 4.32 | nd | na | na |
| CJ26255 | <1.0 | + | 4.45 | 5.54 | na | na |
| CJ26494 | <1.0 | 2.11 | 5.38 | nd | na | na |
| CJ26950 | <1.0 | + | 4.81 | 6.78 | na | na |
| ML29-vaccinated | ||||||
| CJ26493 | <1.0 | 2.67 | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
| CJ27008 | <1.0 | <1.0/- | <1.0/ + | <1.0/- | <1.0/- | <1.0/- |
| CJ26190 | <1.0 | <1.0 | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
| CJ26946 | <1.0 | <1.0/+ | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
| CJ26256 | <1.0 | <1.0/- | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
| CJ26249 | <1.0 | <1.0/- | <1.0/- | <1.0/- | <1.0/- | <1.0/- |
a Viremia in blood was measured by plaque titration and expressed as log10 PFU/ml. Average from two plaque titrations is presented. Viral load is expressed as log10 PFU/ml; <1.0, lower than detection limit; +, sample was positive in co-cultivation assay; <1.0/+, negative in direct plaque assay, but positive in co-cultivation assay; <1.0/-, negative in plaque assay and in co-cultivation assay
A shortage and high cost of rhesus macaques complicated efficacy trials, especially challenge experiments in a BSL-4 environment. To overcome this problem we recently developed a small non-human primate model of LF in the common marmoset, Callitrichid jacchus [84]. Immunization of marmosets with ML29 increased populations of CD14+ cells and CD3+ T lymphocytes in circulating blood. We also found recruitment of CD3+ T cells and over-expression of HLA-DR, P, Q in target tissues [181]. Taken together these data indicate that ML29 immunization resulted in antigen stimulation. In contrast, LASV infection in marmosets was associated with lymphoid depletion, marked reduction in CD3+, CD20+ cells, and down-regulation of class II MHC antigens [84].
A single s.c. inoculation of ML29 vaccine (1000 PFU) induced specific cell-mediated T cell responses assayed in IFN-γ or TNF-α ELISPOT and these responses seem to be responsible for full protection animals against LASV challenge (Figure 4B). No vaccinated animals had any clinical manifestations and all survived the observation period lasting 35 days after challenge. In vaccinated and challenged animals blood chemistry and hematology data did not reveal differences from pre-challenge values (not shown). In contrast, challenge controls showed reduction of platelet numbers, elevated liver enzymes, and decreased levels of albumin in plasma as was previously described [84].
Plasma samples collected after challenge were tested by plaque assay and by co-cultivation on Vero cells. As seen in Table 3, in all non-vaccinated marmosets LASV was detectable on day 5 after challenge and the viremia was more than 105 PFU/ml shortly before or on the day of necropsy. Only in one ML29-vaccinated marmoset was LASV detected by plaque assay on day 5. Two other LASV-positive animals had low transient viremia detectable by Vero co-cultivation. After day 15, no animals had LASV in plasma as judged by biological assays or by nested RT/PCR. In tissues of non-vaccinated animals LASV load was high and varied from 103.3 to 107.6 PFU/g. All tested tissues of ML29-vaccinated animals that were challenged with LASV had no detectable infectious LASV [137].
In all tested animal models, including non-human primates [137,143], replication of ML29 was deeply attenuated, did not induce viremia, or any biochemical/clinical abnormalities. Only immunization at very high doses (106 PFU, 1,000-fold higher then protective dose) resulted in low transient viremia in one of six tested monkeys [143]. Recent studies demonstrated that rhesus macaques infected with Simian Immunodeficiency Virus (SIV) and uninfected monkeys responded similarly to ML29 vaccination, and that none developed signs of arenavirus disease or persistence. Furthermore, 5 of 5 SIV-infected rhesus macaques vaccinated with ML29 animals given a heterologous challenge with a lethal dose of LCMV-WE survived without developing disease signs [182]. It indicates that ML29 is safe and immunogenic in SIV-infected rhesus macaques.
In a surrogate model of LF in rhesus macaques inoculated with viscerotropic WE strain of LCMV [72] early transcriptional changes in blood, mostly in genes involved in complement, IFN, and pro-inflammatory pathways, can discriminate virulent vs. non-virulent infection [183]. We applied the similar approach and showed that gene expression patterns in ML29-exposed human PBMC and control, mock-exposed cells, clustered together confirming safety profile of the ML29 in naïve primates [137,143].
In summary, the ML29 is a promising LASV vaccine candidate that: (i) elicits a strong cross-reactive CMI before challenge; (ii) completely protects guinea pigs and marmosets from LF and this protection is highly suggestive of sterilizing immunity; (iii) provides protection against heterologous LASV strains; (iv) protects fatally infected guinea pigs by treatment at 48 h following infection; and (v) safe and immunogenic in NHP with simian AIDS. Still, risk group classification of MOPV (“guilt by association” [9]) and, consequently, ML29 status as a risk group 3 pathogen, is the major obstacle for further development of this vaccine.
4.5.4. Recombinant Yellow Fever 17D expressing LASV antigens.
There are several arguments to consider Yellow Fever 17D (YF17D) as a vaccine vector for expression of LASV antigens. The YF17D is one of the most efficacious and safe vaccines ever developed with a highly favorable benefit-risk profile and excellent cost-effectiveness. Since 1937 more than 540 million doses have been delivered world-wide and no reversion to wild-type YF virus has been reported. Rare observed adverse effects seem to be associated with host factors and are not due to virus reversion [184,185,186]. However, during the past 20-25 years the absence of effective public health policy, social-ecological factors and vaccine shortage resulted in a resurgence of YF (~ 200,000 cases and 30,000 deaths annually) in South America and Africa. Thirty-two African countries with a total population of 610 million people including more than 219 million urbanites are now considered at risk for YF. The vast majority of cases and deaths take place in sub-Saharan Africa [187], where YF endemic areas are overlapping with LF risk zones [3]. Fisher-Hoch & McCormick, well-recognizable leaders in LASV research, noted that “For the long term, a yellow fever/Lassa fever chimera vaccine for use in EPI in West Africa is a very attractive solution” [188].
Based on outstanding YF17D safety records and recent success in molecular biology of flaviviruses, the genetic backbone of YF17D has been used for construction of chimeric YF17D-based viruses expressing prM and E proteins of closely-related flaviviruses, Japaneses encephalitis (JE), Dengue, and West Nile virus. ChimeriVax™-based vaccines against these flaviviruses are currently undergoing Phase II-III clinical testing (reviewed by [189]). Insertion of short immunogenic peptides into E surface glycoprotein protein did not compromise the morphological structure and attenuated phenotype of YF17D. Recombinant YF17D-based vectors have also been used in the development of promising new vaccines carrying immunogenic epitopes of flavivirus-unrelated pathogens (influenza, malaria, oncogenes) [190,191,192,193,194].
ChimeriVax™ technology is not applicable for cloning large flavivirus-unrelated genes. To generate recombinant YF17D viruses expressing LASV genes we developed technology to clone and express LASV GPC between YF17D E and NS1 genes [144]. In brief, a cDNA fragment encompassing the complete sequences encoding LASV GPC, but lacking the 58 amino acids of the GPC signal sequence to prevent interference with YF polyprotein processing, was fused in frame between the YFV E and NS1 genes. The construct was designed so that LASV GPC protein would be released from the YF17D polyprotein by host signalase. In this construct, the C-terminal 23 hydrophobic amino acids of the YF17D E gene were duplicated downstream of the LASV GPC gene to serve as a signal sequence to ensure insertion of the YFV NS1 protein into the endoplasmic reticulum (ER). In vitro-made RNA from the recombinant clone and from the parental YF17D clone was transfected into BHK-21J cells by electroporation and the recombinant virus was collected from culture medium.
The YF17D/LASV∆GPC recombinant virus was replication-competent, deeply attenuated, induced immune responses against both pathogens, YFV and LASV, and protected 80% of guinea pigs against fatal LF in proof-of-concept challenge experiments [144,195]. In spite of these encouraging results, we [144] and others [196] faced instability problems when large inserts were cloned at the E-NS1 site. It has been suggested that duplication of flavivirus sequences flanking an inserted gene contributed to the genetic instability of the virus. Two approaches were applied to address this problem: (i) insertion of modified fusion sequences between the C-terminal part of foreign inserts and NS1 gene to prevent possible recombination events resulting in deletion of the foreign gene; and (ii) reduction of insert size. We have shown that modification of fusion sequences improved stability but did not preserve the large ∆GPC insert from the deletion during 10 passages in tissue culture. When the size of the insert was reduced almost 2-fold, GP1 and GP2 sub-units of LASV GPC were successfully cloned at the E-NS1 site and these inserts were stable during 10 passages. The size of the LASV GP1 is 200 aa and the size of GP2 is 231 aa residues. In accord with our results, a similar sized insert, 224 aa residues, from SIVmac239 Gag was cloned between the genes encoding the viral proteins E and NS1. The resulting recombinant virus, the YF17D/SIVGag45-269, was stable in vivo and able to induce SIV-specific CD8+ T cell responses in rhesus macaques [197].
The size limitation of foreign gene inserts in YF17D vector seems not to be unique for the E-NS1 site. Recently recombinant YFV17D vaccine was engineered by cloning CSP (aa 57-344) from P. yoelii using a novel insertion site, the capsid gene. The insert was cloned in frame between the highly conserved cyclization sequences (nt 1-75) and FDMV 2A and Ubi genes which were inserted to ensure appropriate cleavage [198]. Notably, the CSP insert was intact up to 6 passages in tissue culture and became undetectable by RT/PCR by passage 7. We have used a similar strategy with limited success in attempts to clone and express overlapping fragments of LASV NP resembling in some ways cytoplasmic YFV core protein (P. Bredenbeek et al; unpublished).
Taken together these results indicate that a monocistronic YF17D genome encoding a relatively large flavivirus-nonrelated gene inserts at the E-NS1 site or in the C gene can produce viable recombinant viruses in cell cultures. However, genetic stability of recombinant viruses is the major problem.. We have shown here that cloning of inserts of moderate size (200-230 aa residues) between the genes encoding the viral proteins E and NS1 resulted in genetically stable recombinant viruses during 10 passages in tissue culture. The YF17D genome itself is remarkably stable [199]. Ten passages of recombinant YFV17D-based viruses will be sufficient to manufacture non-GMP lots for toxicology and pre-clinical studies and for making Phase I lots during vaccine development [189].
Insertion of foreign genes into the YFV17D genome resulted in additional attenuation of chimeric or recombinant YF17D-based viruses. In plasma and tissue of vaccinated guinea pigs [144] and marmosets (I. S. Lukashevich, unpublished) YF17D/LASV-GPC was not detectable by plaque assay and only transient and limited replication was confirmed by co-cultivation in Vero and/or by RT/PCR in line with previously published data [189,200,201]. Still, we were able to detect anti-YFV antibodies in YFV17D/LASV∆GPC-immunized animals. Immunization with recombinant YFV17D/LASV∆GPC or combined immunization with YF17D/LASV-GP1 and -GP2 induced the same levels of protection in strain 13 guinea pigs. A single-dose immunization (1x105-1x106 PFU) or prime-boost immunization (on day 14 or 30 with the same doses) and challenge on day 30 after immunization with 1000 PFU of LASV (Jos) protected 80% of strain 13 guinea pigs against fatal LF [144,145]. However, vaccination of common marmosets with recombnant YF17DV expressing LASV∆GPC using the same protocol did not induce protective immune responses. All vaccinated marmosets died with LF clinical manifestations as in control, no vaccine group (I. S. Lukashevich, unpublished). While safety is not a concern for recombinant YFV17D/LAS vaccines, poor immunogenicity would require additional development before it could be a viable option as a bivalent vaccine to control both infections in overlapping endemic areas in Africa.
5. Current challenges and problems to overcome
5.1. The FDA Animal Rule
The FDA Animal Rule states that under specific circumstances, when human trials would be unethical and unfeasible, the FDA may grant marketing approval to a new vaccine following efficacy trials in adequate and well-controlled animal studies [177]. Whereas pre-clinical toxicity and efficacy studies under the Animal Rule are the same as for clinical trial products, efficacy studies must be performed in more than one well-established and developed animal models. It means to qualify for approval under the Animal Rule, at least two animal models mimicking the pathophysiological mechanisms of human LF must be established in which correlates of protection must be clearly defined.
The NHP are the only relevant model for human LF. Closely-related rhesus (Macaca mulata) and cynomolgous (Macaca fascicularis) monkeys have been most extensively used for evaluation of promising vaccine candidates or treatment [35,76,77,140,142,202,203,204]. In most of these studies, necropsy was performed at the termination stage of the disease. Nevertheless, recent studies focusing on early stages of the LF in NHP [205] and immune responses [82] confirmed previous observations and reestablish markers of fatal LF: unchecked viremia, elevated liver enzymes, low or undetectable levels of proinflammatory cytokines (IL-1β, TNF-α, IL-8, and IP-10), and low and/or ineffective T cell activation. In addition, high levels of IL-6 was found as an additional biological marker of fatal disease linked to hepatocyte regeneration, which had been previously described in fatally infected LF patients [65] and recently confirmed in our surrogate model of LASV-induced hepatitis in rhesus macaques [80,81]. These studies also showed that early and strong CMI responses were associated with effective control of virus replication and recovery.
Efficacy studies in NHP in BS-4 containment are extremely costly. In addition, there is currently a shortage of rhesus macaques for biomedical research [206]. The development of less expensive and more reliable models of human LF for LASV vaccine research is therefore warranted. The common marmoset, Callithrix jacchus, is a small anthropoid primate that generally weighs between 320 to 450 g. The relatively small size of marmosets translates to lower cage and feeding costs, and eases handling in a biosafety environment; these features confer substantial benefits when compared with the use of macaques. Completion of sequence analysis of the entire common marmosets genome will clarify the genetic similarity between the marmosets and humans and will provide access to reliable immunological tools. Because of these advantages, common marmosets have been widely used in many studies involving gene therapy, bacterial infection, toxicology, immunology, and vaccine development [206,207,208,209].
Experimental infection of marmosets with LASV resulted in a systemic disease with high viremia and viral RNA load in tested tissues, elevated liver enzymes, decreased plasma albumin, weight loss, and severe morbidity, the latter which manifests 15 to 20 days after inoculation [84]. Morphological features mirror those described for human cases of fatal LF, and includes hepatic and adrenal necrosis, lymphoid depletion, and interstitial nephritis. Immunochemistry studies of liver and lymphoid tissues revealed marked reduction in CD3+, CD20+ cells, the intensity of HLA-DP, DQ, DR staining, and expression of MHC class II molecules. These observations provided the first experimental evidence that replication of LASV in tissues is associated with immunological alterations that reflect an impaired adaptive immune responses [84]. Common marmosets have been successfully used to evaluate safety, immunogenicity, and efficacy of the LASV ML29 vaccine candidate [143] (see section 4.5.3), providing evidence that LASV infection of marmosets corresponds well with the human LF, like the macaques. Challenge studies in marmosets will considerably reduce cost, especially if breeding colonies are established in-house. LASV infection of marmosets could therefore be the 2nd “small” NHP model to comply with the FDA Animal Rule.
Under current regulation policy, it is highly unlikely that experimental rodents, even the highly susceptible strain 13 guinea pigs, will be considered as an appropriate model for efficacy trials. That is also true for model systems based on infection with host-adapted LASV strains [210] or with less pathogenic “surrogate” arenaviruses (rev. [208]). Recently animal models based on mice with artificially compromised immune system have been described (e.g; mice expressing humanized MHC class I molecules [211], mice deficient for α/β and γ-interferon receptors [212]). These animals can potentially be helpful for basic studies, but probably will not be applicable for efficacy trials.
Although mice do not accurately model human LF disease, they can provide an economical assay to determine vaccine potency via the capacity of vaccine candidates to elicit protective CMI responses. This type of small animal models is especially needed when promising vaccine technology will be transferred from the laboratory to the manufacturing environment. An additional benefit afforded by murine models is a lower level of biocontainment. This level must be lower than BSL-4, because pre-clinical vaccine development in the BSL-4 is not practical. In a recently described CBA/J-ML29 model [178], a T cell cytotoxicity assay in vivo showed a correlation between LASV-specific cytotoxicity and protection induced by the LASV vaccine candidate (ML29). Notably, CBA/J mice that received CD8+ T cell-depleted splenocytes from ML29-immunized donors all succumbed to a lethal i.c. challenge, demonstrating that CD8+ T cells are critical in protection. The CBA/J-ML29 model can therefore be a useful immunological tool for evaluation of immunogenicity and efficacy of LASV vaccine candidates outside of BSL-4 containment facilities.
While results of safety and efficacy studies in NHP models must be submitted to FDA, the agency will probably require efficacy trials in humans. The Animal Rule pertains to diseases which are so rare that trials are not feasible (e.g; outbreaks of hemorrhagic fevers caused by EBOV and MARV). In contrast, LF is common disease in West Africa and this argument cannot be made. Indeed, efficacy evaluation of Candid #1 was performed in phase III trial in Argentina [138]. Notably, prevalence of JUNV infection in Argentina is lower than LASV infection in West Africa; 3,500 cases/yr (historical high before introduction of Candid #1) vs. 3,000-5,000/yr cases of LF in West Africa (conservative estimates). Due to strain variations of LASV, a multicenter trial in several endemic areas (countries) would probably have to be conducted to evaluate cross-protective efficacy of vaccine candidates.
5.2. Vaccine formulation and target population
Vaccine formulation will probably depend on the vaccination strategy and targeted groups. For general population in LASV endemic areas a single-dose vaccination providing a life-long protection against the disease caused by all genetic clades is the most desirable strategy. A live attenuated vaccine expressing LASV GPC and NP would be an appropriate vaccine candidate to meet this goal. LASV NP is the major component of transcription/replication machinery and this protein is synthesized at early stages of infection, prior to GPC and structural glycoproteins. Thus, effective anti-NP immunity during early stages of the infection will potentially contain and control virus replication. Indeed, mice that were tolerated against all NP-derived T cell epitopes were severely compromised in their ability to control LCMV [213]. These findings are in-line with data from humans, in which asymptomatic, but LASV-seropositive, individuals from endemic areas have very strong memory CD4+ T-cell responses against the NP. Taken together, these findings indicate that anti-NP immunity is involved in efficient virus control and contribute to long-term cross-reactivity [61]. Reassortant ML29 is currently available preventive vaccine candidate inducing sterilizing, cross-protective immunity and acting as an effective post-exposure countermeasure. Recently established LASV reverse genetics systems [158,214] provide powerful tools to insure safety profile and further improvement of this vaccine.
Recombinant VSV∆G-based vaccine candidates that express filovirus glycoproteins were intensively tested in more than 80 NHPs with very encouraging results that provide convincing evidence of the safety of this platform (rev. [150,215]). These results justify further development of VSV∆G as a vaccine platform for human use. Unfortunately, the available information on VSV∆G/LASVGP vaccine is very limited. The mechanism of protection is also not clearly understood and durability of protective immunity is unknown. The cross-protective activity of this vaccine against heterogeneous LASV strains must also be addressed. As a possible solution to this concern, vaccination with a blended vaccine that contains equal amounts of rVSV expressing LASV GPC derived from all LASV clades can be considered.
IMOJEV®, a live-attenuated vaccine obtained by replacing the envelope genes of YF-17D with those of JE is expected to become the first approved human viral vector vaccine [216]. Tetravalent dengue vaccine (Sanofi Pasteur) is currently in phase III of clinical development [217]. The success of YF17D-based vaccine technology provides additional motivation to further develop and improve the YF17D/LASV-GPC vaccine platform. This vaccine can be potentially used as a pediatric bivalent vaccine to control both YF and LF infections in overlapping endemic areas [9]. In spite of positive proof-of-concept results in guinea pigs, the poor immunogenicity of this vaccine failed to protect NHPs. Certainly, additional development efforts are required to improve genetic stability of LASV GP inserts and immunogenicity of this vaccine [128].
The advanced VEEV TC-83 replicon system provides a safe and multivalent solution for making effective LASV vaccine candidates. This platform requires a prime-boost vaccination strategy to achieve desirable levels of immune responses and protection. In practical terms, this strategy would be applicable for “organized” target groups (e.g; first responders, personnel of local hospitals in endemic areas, international travelers visiting endemic areas, military personnel, and staff of BSL4 labs working with LASV). The ability of VEEV RPs to deliver and express at least two foreign genes in target cells [96] provides a good opportunity to make cross-protective vaccines by expressing LASV GPC from different clades and with enhanced immunogenic capacities.
5.3. Conclusions and prospects for future
During the past two decades, biodefense funding has brought new human and financial resources to combat emerging infectious diseases, and resulted in substantial progress in basic research (discovery of new arenaviruses, host cell interaction, reverse genetics, etc.). Although progress in vaccine development has been less impressive, at least three vaccine platforms (recombinant vaccinia, MOP/LAS reassortant, and recombinant VSV) provided vaccine candidates that were successful when tested in NHPs. Vaccinia virus is no longer acceptable in Africa with high prevalence of HIV-1, but MOP/LAS reassortant (ML29) and rVSV vaccines have some room for further development, in spite of safety issues surrounding their potential use. The rVSV-based vaccines against filoviruses (EBOV, MARV) have been extensively tested in NHPs with promising results. While filoviruses brought much more public attention and resources from their high mortality and concern over their potential use as bioweapons, they are a relatively minor public health threat for Africa. LASV is a potential bioterrorism threat as well; however, in contrast to filoviruses, LF is also a major public health concern across West Africa [218]. Any progress in clinical development of recombinant VSV-based filovirus vaccines, for example, pre-investigational new drug (IND) approval, will be a step in the right direction [150] and will likely positively impact VSV∆G/LASVGP development as well.
While health authorities of endemic and non-endemic countries have to apply the internationally recognizable regulatory standards to new preventive and therapeutic biologics against emerging infectious threats, it seems that regulatory priorities differ. In the absence of market-driving forces, the approval process in non-endemic countries is very slow. The FDA Animal Rule for regulation of “biodefense” biologics was proposed at the end of the last century and was implemented in 2002. To date, there are no FDA-approved vaccines using the Animal Rule. Meanwhile, in 2007 a live-attenuated vaccine Candid #1, with a clinically proved efficacy of 95% against Argentine hemorrhagic fever , was incorporated in National Immunization Plan in Argentina and tremendously impacted the magnitude of epidemic outbreaks [138,219]. This vaccine was jointly developed by Argentine and US scientists, with support from the national Ministry of Health and US Army Medical Research Institute of Infectious Diseases, under supervision of United Nations Development Program and Pan American Health Organization. In August 2006, after successful clinical trials, the vaccine was registered by the National Regulatory Authorities and is currently manufactured in Argentina. In the U.S. this vaccine has an IND status. It is already made a positive impact on JUNV research. Based and the availability of an effective vaccine, CDC and NIH allowed research with JUNV in BSL3 containment, under the conditions that personnel are vaccinated with Candid #1 [220]. Nevertheless, taking into consideration an apparent negative bias of the FDA regarding replicative-competent vaccines, the feasibility of approval for use in the US is murky. There is a chance that Candid #1 will share the VEEV TC-83 vaccine story. This IND vaccinehas been used for decades to protect military and lab personnel, but it will be never approved by FDA. Nevertheless, so far the Candid #1 story is a promising guide for ML29 vaccine candidate. The major lesson learned from the past is that local needs and responsiveness of national authorities from endemic areas must be important driving forces supported by international development programs and charitable organizations. Recent international initiatives [2] and signs of interest from biotechnology companies [102] provide some hope for the future.
Acknowledgments
The author’s Lassa virus vaccine research was supported by grants R01AI052367, R01AI068961, and R01AI093450 from the National Institutes of Health. The author would like to thank Dr. Gavin Arteel for helpful discussions and editing to improve the manuscript; Dr. E. Fichet-Calvet and Dr. A. Ishii for permission to use their results for Figure 1; and Juan Zapata for help with Figure 3.
Conflict of Interest
The author declares no conflict of interest.
References
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa fever. Curr. Top. Microbiol. Immuno.l 2002, 262, 75–109. [Google Scholar]
- Khan, S.H.; Goba, A.; Chu, M. New opportunities for field research on the pathogenesis and treatment of Lassa fever. Antivir. Res. 2008, 78, 103–115. [Google Scholar]
- Fichet-Calvet, E.; Rogers, D.J. Risk Maps of Lassa Fever in West Africa. PLoS Negl. Trop. Dis. 2009, 3. [Google Scholar]
- Loureiro, M.E.; Wilda, M.; Levingston-Macleod, J.M. Molecular determinants of Arenavirus Z protein homo-oligomerization and L polymerase binding. J. Virol. 2011. JVI.05691-05611.. [Google Scholar]
- Richmond, J.K.; Baglole, D. Lassa fever: epidemiology, clinical features, and social consequences. BMJ 2003, 327, 1271–1275. [Google Scholar] [CrossRef]
- Safronetz, D.; Job, E.; Nafomon, S.; Traore, S.F.; Raffel, S.J.; Fischer, E.R.; Ebihara, H.; Branco, L.; Garry, R.F.; Schwan, T.G.; Feldmann, H. Detection of Lassa Virus. Mali. Emerg. Infect. Dis. 2010, 16, 1123–1126. [Google Scholar] [CrossRef]
- Andersen, K.G.; Shylakhter, I.; Tabrizi, S.; Grossman, S.R.; Happi, C.T.; Sabeti, P.C. Genome-wide scans provide evidence for positive selection of genes implicated in Lassa fever. Philos. T. R. Soc. B. 2012, 367, 868–877. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Varilly, P.; Fry, B. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar]
- Fisher-Hoch, S.P.; McCormick, J.B. Lassa fever vaccine: A review. Expert. Rev. Vaccines 2004, 3, 103–111. [Google Scholar]
- Ishii, A.; Thomas, Y.; Moonga, L.; Nakamura, I.; Ohnuma, A.; Hang'ombe, B.; Takada, A.; Mweene, A.; Sawa, H. Novel arenavirus, Zambia. Emerg. Infect. Dis. 2011, 17, 1921–1924. [Google Scholar] [CrossRef]
- Salvato, M.S.; Clegg, J.C.S.; Buchmeier, M.J. Arenaviridae. In Virus Taxonomy, VIIIth Report of the ICTV; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier/Academic Press: London, UK, 2005; pp. 725–733. [Google Scholar]
- Lukashevich, I.S.; Salvato, M.S. Lassa Virus Genome. Current Genomics 2006, 7, 351–379. [Google Scholar]
- Salvato, M.S.; Clegg, J.C.S.; Buchmeier, M.J. Arenaviridae. In Virus Taxonomy, VIIIth Report of the ICTV; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier/Academic Press: London, UK, 2005; pp. 725–733. [Google Scholar]
- Salvato, M.S.; Clegg, J.C.S.; Buchmeier, M.J.; Charrel, R.N.; Gonzalez, J.P.; Lukashevich, I.S.; Peters, C.J.; Romanowski, V. Family Arenaviridae, Virus Taxonomy, Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses; Andrew King, M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Academic Press, Elsevier Inc.: Amsterdam, The Netherlands, 2012; pp. 715–723. [Google Scholar]
- Charrel, R.N.; de Lamballerie, X.; Emonet, S. Phylogeny of the genus Arenavirus. Curr. Opin. Microbiol. 2008, 11, 362–368. [Google Scholar]
- Emonet, S.F.; de la Torre, J.C.; Domingo, E.; Sevilla, N. Arenavirus genetic diversity and its biological implications. Infect. Genet. Evol. 2009, 9, 417–429. [Google Scholar] [CrossRef]
- Charrel, R.N.; Lemasson, J.J.; Garbutt, M. New insights into the evolutionary relationships between arenaviruses provided by comparative analysis of small and large segment sequences. Virology 2003, 317, 191–196. [Google Scholar]
- Bowen, M.; Rollin, P.; Ksiazek, T. Genetic diversity among Lassa virus strains. J. Virol. 2000, 74, 6992–7004. [Google Scholar] [CrossRef]
- Lozano, M.E.; Posik, D.M.; Albarino, C.G. Characterization of arenaviruses using a family-specific primer set for RT-PCR amplification and RFLP analysis. Its potential use for detection of uncharacterized arenaviruses. Virus Res. 1997, 49, 79–89. [Google Scholar] [CrossRef]
- Bowen, M.D.; Peters, C.J.; Nichol, S.T. Phylogenetic analysis of the Arenaviridae: patterns of virus evolution and evidence for cospeciation between arenaviruses and their rodent hosts. Mol. Phylogenet. Evol. 1997, 8, 301–316. [Google Scholar]
- Gunther, S.; Emmerich, P.; Laue, T. Imported Lassa fever in Germany: molecular characterization of a new Lassa virus strain. Emerg. Infect. Dis. 2000, 6, 466–476. [Google Scholar] [CrossRef]
- Vieth, S.; Torda, A.E.; Asper, M.; Schmitz, H.; Gunther, S. Sequence analysis of L RNA of Lassa virus. Virology 2004, 318, 153–168. [Google Scholar] [CrossRef]
- Emonet, S.; Lemasson, J.J.; Gonzalez, J.P.; Lamballerie, X.d.; Charrel, R.N. Phylogeny and evolution of old world arenaviruses. Virology 2006, 350, 251–257. [Google Scholar]
- Clegg, J. Molecular phylogeny of the Arenaviridae. Curr. Top. Microbiol. Immunol. 2002, 262, 1–24. [Google Scholar] [CrossRef]
- Lalis, A.; Leblois, R.; Lecompte, E.; Denys, C.; ter Meulen, J.; Wirth, T. The Impact of Human Conflict on the Genetics of Mastomys natalensis and Lassa Virus in West Africa. PLoS One 2012, 7, e37068. [Google Scholar]
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa fever. Curr. Top. Microbiol. Immunol. 2002, 262, 75–109. [Google Scholar]
- Peters, C.J.; Jahrling, P.B.; Liu, C.T.; Kenyon, R.H.; McKee, K.T.; Oro, J.G.B. Experimental studies of arenaviral hemorrhagic fevers. Curr. Top. Microbiol. Immunol. 1987, 134, 5–68. [Google Scholar]
- Swanepoel, R.; Leman, P.A.; Shepherd, A.J.; Shepherd, S.P.; Kiley, M.P.; McCormick, J.B. Identification of Ippy as a Lassa-fever-related virus. Lancet 1985, 1, 639. [Google Scholar]
- Meunier, D.Y.; McCormick, J.B.; Georges, A.J.; Georges, M.C.; Gonzalez, J.P. Comparison of Lassa, Mobala, and Ippy virus reactions by immunofluorescence test. Lancet. 1985, 1, 873–874. [Google Scholar]
- Gonzalez, J.; McCormick, J.B.; Saluzzo, J.F.; Herve, J.P.; Georges, A.J.; Johnson, K.M. An arenavirus isolated from wild-caught rodents (Pramys species) in the Central African Republic. Intervirol. 1983, 19, 105–112. [Google Scholar] [CrossRef]
- Wulff, H.B.M.; McIntosh, D.B.; Hamner & Johnson, K.M. Isolation of an arenavirus closely related to lassa virus from rodents. Bull. WHO. 1977, 55, 441–444. [Google Scholar]
- Johnson, K.M.; Taylor, P.; Elliott, L.H.; Tomori, O. Recovery of a Lassa-Related Arenavirus in Zimbabwe. Am. J. Trop. Med. Hyg. 1981, 30, 1291–1293. [Google Scholar]
- Georges, A.; Gonzalez, J.P.; Abdul-Wahid, S.; Saluzzo, J.F.; Meunier, D.M.; McCormick, J.B. Antibodies to Lassa and Lassa-like viruses in man and mammals in the Central African Republic. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 78–79. [Google Scholar] [CrossRef]
- Kiley, M.P.; Lange, J.V.; Johnson, K.M. Protection of rhesus monkeys from Lassa virus by immunisation with closely related Arenavirus. Lancet 1979, 2, 738. [Google Scholar]
- Walker, D.H.; Johnson, K.M.; Lange, J.V.; Gardner, J.J.; Kiley, M.P.; McCormick, J.B. Experimental infection of rhesus monkeys with Lassa virus and a closely related arenavirus, Mozambique virus. J. Infect. Dis. 1982, 146, 360–363. [Google Scholar]
- Fisher-Hoch, S.P.; McCormick, J.B. Lassa fever vaccine. Expert Rev. Vaccines 2004, 3, 103–111. [Google Scholar]
- Salazar-Bravo, J.; Ruedas, L.A.; Yates, T.L. Mammalian reservoirs of arenaviruses. Curr. Top. Microbiol. Immunol. 2002, 262, 25–63. [Google Scholar]
- Keenlyside, R.A.; McCormick, J.B.; Webb, P.A.; Smith, E.; Elliott, L.; Johnson, K.M. Case-control study of Mastomys natalensis and humans in Lassa virus-infected households in Sierra Leone. Am. J.Trop. Med. Hyg. 1983, 32, 829–837. [Google Scholar]
- Demby, A.H.; Inapogui, A.; Kargbo, K.; Koninga, J.; Kourouma, K.; Kanu, J.; Coulibaly, M.; Wagoner, K.D.; Ksiazek, T.G.; Peters, C.J.; Rollin, P.E.; Bausch, D.G. Lassa fever in Guinea: II. Distribution and prevalence of Lassa virus infection in small mammals. Vector Borne Zoonotic Dis. 2001, 1, 283–297. [Google Scholar] [CrossRef]
- Demartini, J.; Green, D.E.; Monath, T.P. Lassa virus infection of Mastomy natalensis in Sierra Leone. Gros and microscopical findings in infected and uninfected animals. Bull World Health Organ 1975, 52, 651–663. [Google Scholar]
- Lecompte, E.; Meulen, J.T.; Emonet, S.; Daffis, S.; Charrel, R.N. Genetic identification of Kodoko virus, a novel arenavirus of the African pigmy mouse (Mus Nannomys minutoides) in West Africa. Virology 2007, 364, 178–183. [Google Scholar] [CrossRef]
- Paweska, J.; Sewlall, N.H.; Ksiazek, T.G.; Blumberg, L.H.; Hale, M.J.; Lipkin, W.I. Nosocomial outbreak of novel arenavirus infection, southern Africa. Emerg. Infect. Dis. 2009, 15, 1598–1602. [Google Scholar]
- Briese, T.; Paweska, J.T.; McMullan, L.K. Genetic Detection and Characterization of Lujo Virus, a New Hemorrhagic Fever-Associated Arenavirus from Southern Africa. PLoS. Pathog. 2009, 4, e1000455. [Google Scholar]
- Palacios, G.; Savji, N.; Hui, J. Genomic and phylogenetic characterization of Merino Walk virus, a novel arenavirus isolated in South Africa. J. Gen. Virol. 2010, 91, 1315–1324. [Google Scholar] [CrossRef]
- Coulibaly-N'Golo, D.; Allali, B.; Kouassi, S.K. Novel Arenavirus Sequences in Hylomyscus sp. and Mus (Nannomys) setulosus from Co´te d'Ivoire: Implications for Evolution of Arenaviruses in Africa. PLoS ONE 2011, 6, e20893. [Google Scholar] [CrossRef]
- Stenglein, M.D.; Sanders, C.; Kistler, A.L. Identification, Characterization, and In Vitro Culture of Highly Divergent Arenaviruses from Boa Constrictors and Annulated Tree Boas: Candidate Etiological Agents for Snake Inclusion Body Disease. mBio 2012, 3. [Google Scholar]
- Kerber, R.; Rieger, T.; Busch, C. Cross-species analysis of the replication complex of Old World arenaviruses reveals two sites in nucleoprotein involved in L protein function. J. Virol. 2011, 85, 12518–12528. [Google Scholar]
- Lukashevich, I.S. Generation of reassortants between African arenaviruses. Virology 1992, 188, 600–605. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; A.D. Vasiuchkov, T.A.; Stel'makh, E.P.; Scheslenok & Shabanov, A.G. The isolation and characteristics of reassortants between the Lassa and Mopeia arenaviruses. Vopr. Virusol. 1991, 36, 146–150. [Google Scholar]
- Cummins, D.; McCormick, J.; Bennett, D. Acute sensorineural deafness in Lassa fever. JAMA. 1990, 264, 2093–2096. [Google Scholar] [CrossRef]
- Kitching, A.; Addiman, S.; Cathcart, S. A fatal case of Lassa Fever in London, January 2009. EUROSURVEI L LANCE 2009, 14, 1–3. [Google Scholar]
- Moraz, M.-L.; Kunz, S. Pathogenesis of arenavirus hemorrhagic fevers. Expert Review of Anti-infective Therapy 2011, 9, 49–59. [Google Scholar] [CrossRef]
- Baize, S.; Pannetier, D.; Faure, C.; Marianneau, P.; Marendat, I.; Georges-Courbot, M.C.; Deubel, V. Role of interferons in the control of Lassa virus replication in human dendritic cells and macrophages. Microbes Infect. 2006, 8, 1194–1202. [Google Scholar]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.C.; Deubel, V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar]
- Baize, S.; Marianneau, P.; Loth, P. Early and strong immune responses are associated with control of viral replication and recovery in Lassa virus-infected cynomolgus monkeys. J. Virol. 2009. JVI.01948-01908.. [Google Scholar]
- Hayes, M.W.; Carrion, R.; Nunneley, J.; Medvedev, A.E.; Salvato, M.S.; Lukashevich, I.S. Pathogenic Old World Arenaviruses Inhibit TLR2/Mal-Dependent Pro-Inflammatory Cytokines in vitro. J. Virol. 2012, 86, 7216–7226. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Maryankova, R.F.; Vladyko, A.S. Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: different effects on IL-8 and TNF-a gene expression. J. Med. Virol. 1999, 59, 552–560. [Google Scholar] [CrossRef]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar] [CrossRef]
- Mahanty, S.; Hutchinson, K.; Agarwal, S.; Mcrae, M.; Rollin, P.E.; Pulendran, B. Cutting Edge: Impairment of Dendritic Cells and Adaptive Immunity by Ebola and Lassa Viruses. J. Immunol. 2003, 170, 2797–2801. [Google Scholar]
- Meulen, J.; Badusche, M.; Satoguina, J. Old and New World arenaviruses share a highly conserved epitope in the fusion domain of the glycoprotein 2, which is recognized by Lassa virus-specific human CD4+ T-cell clones. Virology 2004, 321, 134–143. [Google Scholar]
- Ter Meulen, J.; Badusche, M.; Kuhnt, K. Characterization of human CD4+ T-cell clones recognizing conserved and variable epitopes of the Lassa virus nucleoprotein. J. Virol. 2000, 74, 2186–2192. [Google Scholar]
- Jahrling, P.B.; Frame, J.D.; Smith, S.B.; Monson, M.H. Endemic Lassa fever in Liberia. III. Characterization of Lassa virus isolates. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 374–379. [Google Scholar] [CrossRef]
- Walker, D.; McCormick, J.B.; Johnson, K.M.; Webb, P.A.; Komba-Kono, G.; Elliott, L.H.; Gardner, J.J. Pathologic and virologic study of fatal Lassa fever in man. Am. J. Pathol. 1982, 107, 349–356. [Google Scholar]
- Knobloch, J.M.J.; Webb, P.A.; Dietrich, M.; Schumacher, H.H.; Dennis, E. Clinical observations in 42 patients with Lassa fever. Tropenmed Parasitol. 1980, 31, 389–398. [Google Scholar]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Elliott, L.H.; Whitfield, S.G.; Johnson, K.M. Lassa virus hepatitis: a study of fatal Lassa fever in humans. Am. J. Trop. Med. Hyg. 1986, 35, 401–407. [Google Scholar]
- Geisberg, T.W.; Jahrling, P.B. Exotic emerging viral diseases: progress and challenges. Nature Medicine 2004, 10, 110–S121. [Google Scholar]
- Ter Meulen, J.; Lukashevich, I.S.; Sidibe, K. Hunting of peridomestic rodents and consumption of their meat as possible risk factors for rodent-to-human transmission of Lassa virus in the Republic of Guinea. Am. J. Trop. Med. Hyg. 1996, 55, 661–666. [Google Scholar]
- Dylla, D.E.; Michele, D.E.; Campbell, K.P.; McCray, P.B., Jr. Basolateral Entry and Release of New and Old World Arenaviruses from Human Airway Epithelia. J. Virol. 2008, 82, 6034–6038. [Google Scholar]
- Schlie, K.; Maisa, A.; Freiberg, F.; Groseth, A.; Strecker, T.; Garten, W. Viral Protein Determinants of Lassa Virus Entry and Release from Polarized Epithelial Cells. J. Virol. 2010, 84, 3178–3188. [Google Scholar] [CrossRef]
- Kunz, S. Receptor binding and cell entry of Old World arenaviruses reveal novel aspects of virus-host interaction. Virology 2009, 387, 245–249. [Google Scholar] [CrossRef]
- Cosset, F.-L.; Marianneau, P.; Verney, G. Characterization of Lassa Virus Cell Entry and Neutralization with Lassa Virus Pseudoparticles. J. Virol. 2009, 83, 3228–3237. [Google Scholar]
- Lukashevich, I.S.; Djavani, M.; Rodas, J.D. Hemorrhagic fever occurs after intravenous, but not after intragastric, inoculation of rhesus macaques with lymphocytic choriomeningitis virus. J. Med. Virol. 2002, 67, 171–186. [Google Scholar] [CrossRef]
- McCormick, J.B. Lassa fever. In Factors in Emergence and Control of Rodent-borne Viral Diseases (Hantaviral and Arenal Diseases) 1999; Saluzzo, J.F., Dodet, B., Eds.; Elsevier: Amsterdam, The Netherlands, 1999; pp. 177–195. [Google Scholar]
- McCormick, J.B.; King, I.J.; Webb, P.A. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986, 314, 20–26. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P. Arenavirus pathophysiology. In The Arenaviridae; Salvato, M.S., Ed.; Plenum Press, 1993; pp. 299–323. [Google Scholar]
- Callis, R.T.; Jahrling & DePaoli, P.B. A Pathology of Lassa virus infection in the rhesus monkey. Am. J. Trop. Med. Hyg. 1982, 31, 1038–1045. [Google Scholar]
- Jahrling, P.B.; Hesse, R.A.; Eddy, G.A.; Johnson, K.M.; Callis, R.T.; Stephen, E.L. Lassa virus infection of rhesus monkeys: pathogenesis and treatment with ribavirin. J. Infect. Dis. 1980, 141, 580–589. [Google Scholar] [CrossRef]
- Lange, J.V.; Mitchell, S.W.; McCormick, J.B.; Walker, D.H.; Evatt, B.L.; Ramsey, R.R. Kinetic study of platelets and fibrinogen in Lassa virus-infected monkeys and early pathologic events in Mopeia virus-infected monkeys. Am. J. Trop. Med. Hyg. 1985, 34, 999–1007. [Google Scholar]
- Kunz, S. The role of the vascular endothelium in arenavirus haemorrhagic fevers. Thromb. Haemost 2009, 102, 1024–1029. [Google Scholar]
- Lukashevich, I.S.; I. Tikhonov, I.; Rodas, J.D. Arenavirus-mediated liver pathology: acute lymphocytic choriomeningitis virus infection of rhesus macaques is characterized by high-level interleukin-6 expression and hepatocyte proliferation. J. Virol. 2003, 77, 1727–1737. [Google Scholar]
- Lukashevich, I.S.; Rodas, J.D.; Tikhonov, I.I. LCMV-mediated hepatitis in rhesus macaques: WE but not ARM strain activates hepatocytes and induces liver regeneration. Arch. Virol. 2004, 149, 2319–2336. [Google Scholar]
- Baize, S.; Marianneau, P.; Loth, P. Early and Strong Immune Responses Are Associated with Control of Viral Replication and Recovery in Lassa Virus-Infected Cynomolgus Monkeys. J. Virol. 2009, 83, 5890–5903. [Google Scholar]
- Pannetier, D.; Reynard, S.; Russier, M. Human Dendritic Cells Infected with the Nonpathogenic Mopeia Virus Induce Stronger T-Cell Responses than Those Infected with Lassa Virus. J. Virol. 2011, 85, 8293–8306. [Google Scholar] [CrossRef]
- Carrion, R.; Brasky, K.; Mansfield, K. Lassa Virus Infection in Experimentally Infected Marmosets: Liver Pathology and Immunophenotypic Alterations in Target Tissues. J. Virol. 2007, 81, 6482–6490. [Google Scholar] [CrossRef]
- Zapata, J.C.; Pauza, C.D.; Djavani, M.M. Lymphocytic choriomeningitis virus (LCMV) infection of macaques: A model for Lassa fever. Antivir. Res. 2011, 92, 125–138. [Google Scholar]
- Zhou, S.; Kurt-Jones, E.A.; Mandell, L.; Cerny, A.; Chan, M.; Golenbock, D.T.; Finberg, R.W. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 2005, 35, 822–830. [Google Scholar]
- Zhou, S.; Kurt-Jones, E.A.; Cerny, A.M.; Chan, M.; Bronson, R.T.; Finberg, R.W. MyD88 Intrinsically Regulates CD4 T-Cell Responses. J. Virol. 2009, 83, 1625–1634. [Google Scholar] [CrossRef]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B.A. Identification of -dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef]
- Kunz, S.; Rojek, J.M.; Kanagawa, M. Posttranslational Modification of {alpha}-Dystroglycan, the Cellular Receptor for Arenaviruses, by the Glycosyltransferase LARGE Is Critical for Virus Bindin. J. Virol. 2005, 79, 14282–14296. [Google Scholar] [CrossRef]
- Oldstone, M.B.A.; Campbell, K.P. Decoding arenavirus pathogenesis: Essential roles for alpha-dystroglycan-virus interactions and the immune response. Virology 2011, 411, 170–179. [Google Scholar] [CrossRef]
- Imperiali, M.; Spörri, R.; Hewitt, J.; Oxenius, A. Post-translational modification of α-dystroglycan is not critical for lymphocytic choriomeningitis virus receptor function in vivo. J. Gen. Virol. 2008, 89, 2713–2722. [Google Scholar]
- Shimojima, M.; Ströher, U.; Ebihara, H.; Feldmann, H.; Kawaoka, Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J Virol. 2012, 86, 2067–2078. [Google Scholar] [CrossRef]
- Shimojima, M.; Kawaoka, Y. Cell surface molecules involved in infection mediated by lymphocytic choriomeningitis virus glycoprotein. J. Vet. Med. Sci. 2012. [Google Scholar]
- Clegg, J.C.; Lloyd, G. Vaccinia recombinant expressing Lassa-virus internal nucleocapsid protein protects guinea pigs against Lassa fever. Lancet. 1987, 2, 186–188. [Google Scholar] [CrossRef]
- Johnson, K.M.; McCormick, J.B.; Webb, P.A.; Smith, E.S.; Elliot, L.H.; King, I.J. Clinical virology of Lassa fever in hospitalized patient. J. Infect. Dis. 1987, 155, 456–463. [Google Scholar]
- Pushko, P.; Geisbert, J.; Parker, M.; Jahrling, P.; Smith, J. Individual and bivalent vaccines based on alphavirus replicons protect guinea pigs against infection with Lassa and Ebola viruses. J. Virol. 2001, 75, 11677–11685. [Google Scholar]
- McCormick, J.; Mitchell, S.; Kiley, M.; Ruo, S.; Fisher-Hoch, S. Inactivated Lassa virus elicits a non protective immune response in rhesus monkeys. J. Med. Virol. 1992, 31, 1–7. [Google Scholar]
- Lukashevich, I.S.; Clegg, J.C.; Sidibe, K. Lassa virus activity in Guinea: distribution of human antiviral antibody defined using enzyme-linked immunosorbent assay with recombinant antigen. J. Med. Virol. 1993, 40, 210–217. [Google Scholar] [CrossRef]
- Bausch, D.G.; Rollin, P.E.; Demby, A.H. Diagnosis and Clinical Virology of Lassa Fever as Evaluated by Enzyme-Linked Immunosorbent Assay, Indirect Fluorescent-Antibody Test, and Virus Isolation. J. Clin. Microbiol. 2000, 38, 2670–2677. [Google Scholar]
- Branco, L.M.; Grove, J.N.; Boisen, M.L.; Shaffer, J.G.; Goba, A.; Fullah, M.; Momoh, M.; Grant, D.S.; Garry, R.F. Emerging trends in Lassa fever: redefining the role of immunoglobulin M and inflammation in diagnosing acute infection. Virol. J. 2011, 8. [Google Scholar]
- Krasnianskiĭ, V.; Potryvaeva, N.V.; Borisevich, I.V.; Gradoboev, V.N.; Pashanina, T.P.; Pshenichnov, V.A. A trial to produce an inactivated Lassa fever vaccine. Vopr Virusol. 1993, 38(6), 276–279. [Google Scholar]
- Branco, L.; Grove, J.; Geske, F. Lassa virus-like particles displaying all major immunological determinants as a vaccine candidate for Lassa hemorrhagic fever. Virology J. 2010, 7, 279. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. Rev. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2008, 20, 17–22. [Google Scholar] [CrossRef]
- Borrow, P.; Martinez-Sobrido, L.; De la Torre, J.C. Inhibition of the Type I Interferon Antiviral Response During Arenavirus Infection. Viruses 2010, 2, 2443–2480. [Google Scholar] [CrossRef]
- Koyama, S.; Coban, C.; Aoshi, T.; Horii, T.; Akira, S.; Ishii, K.J. Innate immune control of nucleic acid-based vaccine immunogenicity. Expert. Rev. Vaccines. 2009, 8, 1099–10107. [Google Scholar] [CrossRef]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E. Junín Virus Infection Activates the Type I Interferon Pathway in a RIG-I-Dependent Manner. PLoS Negl Trop. Dis. 2012, 6, e1659. [Google Scholar] [CrossRef]
- Habjan, M.; Andersson, I.; Klingström, J. Processing of Genome 5′ Termini as a Strategy of Negative-Strand RNA Viruses to Avoid RIG-I-Dependent Interferon Induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef]
- Walsh, K.; Teijaro, J.R.; Zuniga, E.I.; Welch, M.J.; Fremgen, D.M.; Blackburn, S.D.; von Tiehl, K.F.; Wherry, E.J.; Flavell, R.A.; Oldstone, M.B. Toll-like Receptor 7 Is Required for Effective Adaptive Immune Responses that Prevent Persistent Virus Infection. Cell Host Microbe. 2012, 11, 643–653. [Google Scholar] [CrossRef]
- Macal, M.; Lewis, G.M.; Kunz, S.; Flavell, R.; Harker, J.A.; Zúñiga, E.I. Plasmacytoid Dendritic Cells Are Productively Infected and Activated through TLR-7 Early after Arenavirus Infection. Cell Host Microbe. 2012, 11, 617–630. [Google Scholar] [CrossRef]
- Baz Morelli, A.; Becher, D.; Koernig, S.; Silva, A.; Drane, D.; Maraskovsky, E. ISCOMATRIX: a novel adjuvant for use in prophylactic and therapeutic vaccines against infectious diseases. Journal of Medical Microbiology 2012, 61, 935–943. [Google Scholar] [CrossRef]
- Boesen, A.; Sundar, K.; Coico, R. Lassa Fever Virus Peptides Predicted by Computational Analysis Induce Epitope-Specific Cytotoxic-T-Lymphocyte Responses in HLA-A2.1 Transgenic Mice. Clin. Diagn. Lab. Immunol. 2005, 12, 1223–1230. [Google Scholar]
- Botten, J.; Alexander, J.; Pasquetto, V. Identification of Protective Lassa Virus Epitopes That Are Restricted by HLA-A2. J. Virol. 2006, 80, 8351–8361. [Google Scholar] [CrossRef]
- Botten, J.; Whitton, J.L.; Barrowman, P. A Multivalent Vaccination Strategy for the Prevention of Old World Arenavirus Infection in Humans. J. Virol. 2010, 84, 9947–9956. [Google Scholar] [CrossRef]
- Liu, F.; Feuer, R.; Hassett, D.E.; Whitton, J.L. Peptide vaccination of mice immune to LCMV or vaccinia virus causes serious CD8+ T cell-mediated, TNF-dependent immunopathology. J. Clin. Invest. 2006, 116, 465–475. [Google Scholar]
- Strauss, J.; Strauss, E. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar]
- Vander Veen, R.L.; Hank Harris, D.L.; Kurt, I. Kamrud. Alphavirus replicon vaccines. Animal Health Res. Rev. 2012. [Google Scholar]
- Bergen, M.J.; Pan, C.-H.; Greer, C.E.; Legg, H.S.; Polo, J.M.; Griffin, D.E. Comparison of the Immune Responses Induced by Chimeric Alphavirus-Vectored and Formalin-Inactivated Alum-Precipitated Measles Vaccines in Mice. PLoS ONE 2010, 5, e10297. [Google Scholar]
- Carroll, T.; Matzinger, S.R.; Barro, M.; Fritts, L.; McChesney, M.B.; Miller, C.J.; Johnston, R.E. Alphavirus replicon-based adjuvants enhance the immunogenicity and effectiveness of Fluzone® in rhesus macaques. Vaccine 2011, 29, 931–940. [Google Scholar]
- Thompson, J.; Whitmore, A.C.; Konopka, J.L.; Collier, M.L.; Richmond, E.M.; Davis, N.L. Mucosal and systemic adjuvant activity of alphavirus replicon particles. Proc. Natl. Acad. Sci. U. S. A. 2006, 7, 3722–3727. [Google Scholar]
- Thompson, J.; Nicholson, M.G.; Whitmore, A.C.; Zamora, M.; West, A.; Iwasaki, A. Nonmucosal alphavirus vaccination stimulates a mucosal inductive environment in the peripheral draining lymph node. J. Immunol. 2008, 181, 574–585. [Google Scholar]
- Thompson, J.M.; Whitmore, A.C.; Staats, H.F.; Johnston, R.E. Alphavirus replicon particles acting as adjuvants promote CD8+ T cell responses to co-delivered antigen. Vaccine 2008, 26, 4267–4275. [Google Scholar] [CrossRef]
- Pushko, P.; Parker, M.; Ludwig, G.V.; Davis, N.L.; Johnston, R.E.; Smith, J.F. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 1997, 2, 389–401. [Google Scholar]
- Nelson, E.; Prieto, D.; Alexander, T.G.; Pushko, P. Venezuelan equine encephalitis replicon immunization overcomes intrinsic tolerance and elicits effective anti-tumor immunity to the 'self' tumor-associated antigen, neu in a rat mammary tumor model. Breast Cancer Res. Treat. 2003, 82, 169–183. [Google Scholar] [CrossRef]
- Johnston, R.; Davis, N.; Smith, J.; Pushko, P.; Parker, M.; Ludwig, G. Alphavirus RNA replicon systems. U.S. Patent 7,235,235, 2007. Alphavax IP. [Google Scholar]
- Pushko, P. Vector platforms derived from the alphavirus vaccines. U.S. Patent Application No. 2006/0198854 2006. [Google Scholar]
- Rodriguez-Carreno, M.P.; Nelson, M.S.; Botten, J.; Smith-Nixon, K.; Buchmeier, M.J.; Whitton, J.L. Evaluating the immunogenicity and protective efficacy of a DNA vaccine encoding Lassa virus nucleoprotein. Virology 2005, 335, 87–98. [Google Scholar] [CrossRef]
- Grant-Klein, R.; Altamura, L.A.; Schmaljohn, C.S. Progress in recombinant DNA-derived vaccines for Lassa virus and filoviruses. Virus Res. 2011, 162, 148–161. [Google Scholar]
- Whitton, J.L.; Tishon, A.; Lewicki, H. Molecular analyses of a five-amino-acid cytotoxic T-lymphocyte (CTL) epitope: an immunodominant region which induces nonreciprocal CTL cross-reactivity. J. Virol. 1989, 63, 4303–4310. [Google Scholar]
- Schulz, M.; Aichele, P.; Schneider, R.; Hansen, T.H.; Zinkernagel, R.M.; Hengartner, H. Major histocompatibility complex binding and T cell recognition of a viral nonapeptide containing a minimal tetrapeptide. Eur. J. Immunol. 1991, 21, 1181–1185. [Google Scholar] [CrossRef]
- Djavani, M.; Yin, C.; Xia, L.; Lukashevich, I.S.; Pauza, C.; Salvato, M. Murine immune responses to mucosally delivered Salmonella expressing Lassa fever virus nucleoprotein. Vaccine. 2000, 18, 1543–1554. [Google Scholar] [CrossRef]
- Djavani, M.; Yin, C.; Lukashevich, I.; Rodas, J.; Rai, S.; Salvato, M. Mucosal immunization with Salmonella typhimurium expressing Lassa virus nucleocapsid protein cross-protects mice from lethal challenge with lymphocytic choriomeningitis virus. J. Hum. Virol. 2001, 4, 103–108. [Google Scholar]
- Whitton, J.L.; Sheng, N.; Oldstone, M.B.; McKee, T.A. A "string-of-beads" vaccine, comprising linked minigenes, confers protection from lethal-dose virus challenge. Journal of Virology 1993, 67, 348–352. [Google Scholar]
- Saade, F.; Petrovsky, N. Technologies for enhanced efficacy of DNA vaccines. Expert. Rev. Vaccines 2012, 11, 189–2012. [Google Scholar]
- Lauring, A.S.; Jeremy, O. Raul Andino. Rationalizing the development of live attenuated virus vaccines. Nat. Biotechnol. 2010, 28, 573–579. [Google Scholar]
- McCormick, J.B.; Fisher-Hoch, S.P. Lassa fever. Curr. Top. Microbiol. Immunol. 2002, 262, 75–109. [Google Scholar]
- Lukashevich, I.S.; Patterson, J.; Carrion, R. A live attenuated vaccine for Lassa fever made by reassortment of Lassa and Mopeia viruses. J. Virol. 2005, 79, 13934–13942. [Google Scholar] [CrossRef]
- Ambrosio, A.; Saavedra, M.C.; Mariani, M.A.; Gamboa, G.S.; Maiza, A.S. Argentine hemorrhagic fever vaccines. Human Vaccines 2011, 7, 694–700. [Google Scholar] [CrossRef]
- Auperin, D.D.; Esposito, A.A.; Lange, J.V. Construction of a recombinant vaccinia virus expressing the Lassa virus glycoprotein gene and protection of guinea pigs from a lethal Lassa virus infection. Virus Res. 1988, 9, 233–248. [Google Scholar]
- Fisher-Hoch, S.P.; Hutwagner, L.; Brown, B.; McCormick, J.B. Effective Vaccine for Lassa Fever. J. Virol. 2000, 74, 6777–6783. [Google Scholar]
- Garbutt, M.; Liebscher, R.; Wahl-Jensen, V.; Jones, S.; Möller, P.; Wagner, R.; Volchkov, V.; Klenk, H.D.; Feldmann, H.; Ströher, U. Properties of Replication-Competent Vesicular Stomatitis Virus Vectors Expressing Glycoproteins of Filoviruses and Arenaviruses. J. Virol. 2004, 78, 5458–5465. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jones, S.; Fritz, E.A. Development of a new vaccine for the prevention of Lassa Fever. PLoS Medicine 2005, 2, e183. [Google Scholar] [CrossRef] [Green Version]
- Lukashevich, I.S.; Patterson, J. Safety, immunogenicity, and efficacy of the ML29 vaccine for Lassa fever in small non-human primates. Vaccine 2008, 26, 5246–5254. [Google Scholar]
- Bredenbeek, P.J.; Molenkamp, R.; Spaan, W.J.M. A recombinant Yellow Fever 17D vaccine expressing Lassa virus glycoproteins. Virology 2006, 345, 299–304. [Google Scholar] [CrossRef]
- Jiang, X.; Dalebout, T.J.; Bredenbeek, P.J. Yellow fever 17D-vectored vaccines expressing Lassa virus GP1 and GP2 glycoproteins provide protection against fatal disease in guinea pigs. Vaccine 2011, 29, 1248–1257. [Google Scholar] [CrossRef]
- Morrison, H.G.; Bauer, S.P.; Lange, J.V.; Esposito, J.J.; McCormick, J.B.; Auperin, D.D. Protection of guinea pigs from Lassa fever by vaccinia virus recombinants expressing the nucleoprotein or the envelope glycoproteins of Lassa virus. Virology 1989, 171, 179–188. [Google Scholar]
- Fisher-Hoch, S.P.; McCormick, J.B.; Auperin, D. Protection of rhesus monkeys from fatal Lassa fever by vaccination with a recombinant vaccinia virus containing the Lassa virus glycoprotein gene. Proc. Natl. Acad. Sci. USA 1989, 86, 317–321. [Google Scholar]
- Tani, H.; Shigeru, M.; Yoshiharu, M. Development and applications of VSV vectors based on cell tropism. Front Microbiol. 2011. [Google Scholar]
- Geisbert, T.; Feldmann, H. Recombinant vesicular stomatitis virus-based vaccines against Ebola and Marburg virus infections. J. Infect. Dis. 2011, 204, 1075–1081. [Google Scholar]
- Falzarano, D.; Geisbert, T.W.; Feldmann, H. Progress in filovirus vaccine development: evaluating the potential for clinical use. Expert. Rev. Vaccines. 2011, 10, 63–77. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Jones, S.; Fritz, E.A. Development of a new vaccine for the prevention of Lassa fever. PLoS Med. 2005, 2, 537–545. [Google Scholar]
- Charrel, R.N.; de Lamballerie, X.; Fulhorst, C.F. The Whitewater Arroyo virus: natural evidence for genetic recombination among Tacaribe serocomplex viruses (family Arenaviridae). Virology 2001, 283, 161–166. [Google Scholar] [CrossRef]
- Riviere, Y.; Oldstone, M.B. Genetic reassortants of lymphocytic choriomeningitis virus: unexpected disease and mechanism of pathogenesis. J. Virol. 1986, 59, 363–368. [Google Scholar]
- Bishop, D.H.L.; Beaty, B.J.; Shope, R.E. Recombination and gene coding asignment of bunyaviruses and arenaviruses. Ann. N. Y. Acad. Sci. 1980, 354, 84–106. [Google Scholar] [CrossRef]
- Riviere, Y.R.; Ahmed, P.J.; Southern, M.J.; Buchmeier & Oldstone, M.B. Genetic mapping of lymphocytic choriomeningitis virus pathogenicity: virulence in guinea pigs is associated with the L RNA segment. J. Virol. 1985, 55, 704–709. [Google Scholar]
- Zhang, L.; Marriott, K.A.; Harnis, D.G.; Aronson, J.F. Reassortant analysis of guinea pig virulence of Pichinde virus variants. Virology 2001, 290, 30–38. [Google Scholar]
- Moshkoff, D.; Salvato, M.S.; Lukashevich, I.S. Molecular characterization of a reassortant virus derived from Lassa and Mopeia viruses. Virus Genes 2007, 34, 169–176. [Google Scholar] [CrossRef]
- Albarino, C.G.; Bird, B.H.; Chakrabarti, A.K.; Dodd, K.A.; Erickson, B.R.; Nichol, S.T. Efficient rescue of recombinant Lassa virus reveals the influence of S segment non-coding regions on virus replication and virulence. J. Virol. 2011, 85, 4020–4024. [Google Scholar] [CrossRef]
- Hass, M.; Westerkofsky, M.; Muller, S.; Becker-Ziaja, B.; Busch, C.; Gunther, S. Mutational Analysis of the Lassa Virus Promoter. J. Virol. 2006, 80, 12414–12419. [Google Scholar] [CrossRef]
- Perez, M.; de la Torre, J.C. Characterization of the Genomic Promoter of the Prototypic Arenavirus Lymphocytic Choriomeningitis Virus. J. Virol. 2003, 77, 1184–1194. [Google Scholar]
- Barr, J.N.; Wertz, G.W. Bunyamwera Bunyavirus RNA Synthesis Requires Cooperation of 3'- and 5'-Terminal Sequences. J. Virol. 2004, 78, 1129–1138. [Google Scholar] [CrossRef]
- Lukashevich, I.; Djavani, M.; Shapiro, K. The Lassa fever virus L gene: nucleotide sequence, comparison, and precipitation of a predicted 250 kDa protein with monospecific antiserum. J. Gen. Virol. 1997, 78, 547–551. [Google Scholar]
- Lelke, M.; Brunotte, L.; Busch, C.; Gunther, S. An N-Terminal Region of Lassa Virus L Protein Plays a Critical Role in Transcription but Not Replication of the Virus Genome. J. Virol. 2010, 84, 1934–1944. [Google Scholar] [CrossRef]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 2396–2401. [Google Scholar]
- Qi, X.; Lan, S.; Wang, W. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Emonet, S.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de la Torre, J.C. Identification of Amino Acid Residues Critical for the Anti-Interferon Activity of the Nucleoprotein of the Prototypic Arenavirus Lymphocytic Choriomeningitis Virus. J. Virol. 2009, 83, 11330–11340. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Giannakas, P.; Cubitt, B.; Garcia-Sastre, A.; de La Torre, J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Zuniga, E.I.; Rosario, D.; Garcia-Sastre, A.; de la Torre, J.C. Inhibition of the Type I Interferon Response by the Nucleoprotein of the Prototypic Arenavirus Lymphocytic Choriomeningitis Virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Hastie, K.M.; Bale, S.; Kimberlin, C.R.; Saphire, E.O. Hiding the evidence: two strategies for innate immune evasion by hemorrhagic fever viruses. Curr. Opin. Virol. 2012, 2, 151–156. [Google Scholar]
- Glushakova, S.E.; Omelyanenko, V.G.; Lukashevitch, I.S. The fusion of artificial lipid membranes induced by the synthetic arenavirus 'fusion peptide'. Biochim. Biophys. Acta. 1992, 1110, 202–208. [Google Scholar] [CrossRef]
- Klewitz, C.; Klenk, H.-D.; ter Meulen, J. Amino acids from both N-terminal hydrophobic regions of the Lassa virus envelope glycoprotein GP-2 are critical for pH-dependent membrane fusion and infectivity. J. Gen. Virol. 2007, 88, 2320–2328. [Google Scholar]
- Moshkoff, D.; Salvato, M.S.; Lukashevich, I.S. Molecular characterization of a reassortant virus derived from Lassa and Mopeia viruses. Virus Genes 2006. [Google Scholar]
- Kelley, L.A.; Michael, J.E.S. Protein structure prediction on the Web: a case study using the Phyre server. Nature Protocols 2009, 4, 363–371. [Google Scholar]
- Droniou-Bonzom, M.E.; Reignier, T.; Oldenburg, J.E. Substitutions in the Glycoprotein (GP) of the Candid#1 Vaccine Strain of Junin Virus Increase Dependence on Human Transferrin Receptor 1 for Entry and Destabilize the Metastable Conformation of GP. Journal of Virology 2011, 85, 13457–13462. [Google Scholar]
- Albariño, C.G.; Bird, B.H.; Chakrabarti, A.K. The Major Determinant of Attenuation in Mice of the Candid1 Vaccine for Argentine Hemorrhagic Fever Is Located in the G2 Glycoprotein Transmembrane Domain. Journal of Virology 2011, 85, 10404–10408. [Google Scholar] [CrossRef]
- Glushakova, S.E.; Pyzhik, E.V.; Vasiuchkov, A.D.; Erokhina, I.R.; Mar'iankova, R.F. pH-dependent fusion of eukaryotic cells, caused by arenaviruses, pathogenic and non-pathogenic for humans. Mol Gen Mikrobiol Virusol. 1992, 7-8, 27–31. [Google Scholar]
- Snoy, P.J. Establishing Efficacy of Human Products Using Animals. Veterinary Pathology Online 2010, 47, 774–778. [Google Scholar] [CrossRef]
- Goicocheaa, M.A.; Zapata, J.C.; Bryant, J.; Davis, H.; Salvato, M.S.; Lukashevich, I.S. Evaluation of Lassa virus vaccine immunogenicity in a CBA/J-ML29 mouse model. Vaccine 2012, 30, 1445–1452. [Google Scholar] [CrossRef]
- Barkar, N.D.; Lukashevich, I.S. Lassa and Mozambique viruses: cross protection in experiments on mice and action of immunosuppressants on experimental infections. Vopr. Virusol. 1989, 34, 598–603. [Google Scholar]
- Carrion, R.J.; Patterson, J.L.; Johnson, C. A ML29 reassortant virus protects guinea pigs against a distantly-related Nigerian strain of Lassa virus and can provide sterilizing immunity. Vaccine 2007, 25, 4093–4102. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Carrion, R., Jr.; Salvato, M.S. Safety, immunogenicity, and efficacy of the ML29 reassortant vaccine for Lassa fever in small non-human primates. Vaccine 2008. In Press, Corrected Proof.. [Google Scholar]
- Zapata, J.C.; Poonia, B.; Bryant, J.; Davis, H.; Ateh, E.; George, L.; Crasta, O.; Zhang, Y.; Slezak, T.; Crystal Jaing, C.; Pauza, D.; Goicochea, M.; Moshkoff, D.; Lukashevich, I.S.; Salvato, M.S. An attenuated Lassa vaccine in SIV-infected rhesus macaques does not persist or cause arenavirus disease but does elicit Lassa virus-specific immunity. Virology J 2012. submitted. [Google Scholar]
- Djavani, M.M.; Crasta, O.R.; Zapata, J.C. Early blood profiles of virus infection in a monkey model for Lassa fever. J. Virol. 2007, 81, 7960–7973. [Google Scholar] [CrossRef]
- Hayes, E.B. Acute viscerotropic disease following vaccination against yellow fever. Trans R Soc Trop. Med. Hyg. 2007, 101, 967–971. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Schroeder, B.A.; Miller, E.R. Adverse event reports following yellow fever vaccination. Vaccine 2008, 26, 6077–6082. [Google Scholar]
- Silva, M.L.; Espirito-Santo, L.R.; Martins, M.A. Clinical and Immunological Insights on Severe, Adverse Neurotropic and Viscerotropic Disease following 17D Yellow Fever Vaccination. Clin. Vaccine Immunol. 2010, 17, 118–126. [Google Scholar]
- Galbraith, S.E.; Barrett, A.D.T. Vaccines for Biodefense and Emerging and Neglected Diseases. In Yellow Fever; Barrett, A.D.T., Stanberry, L.R., Eds.; Academic Press: Elsevier, Amsterdam, The Netherlands, 2009; pp. 753–785. [Google Scholar]
- Fisher-Hoch, S.; McCormick, J. Towards a human Lassa fever vaccine. Rev. Med. Virol. 2001, 11, 331–341. [Google Scholar] [CrossRef]
- Guy, B.; Guirakhoo, F.; Barban, V.; Higgs, S.; Monath, T.P.; Lang, J. Preclinical and clinical development of YFV 17D-based chimeric vaccines against dengue, West Nile and Japanese encephalitis viruses. Vaccine 2010, 28, 632–649. [Google Scholar] [CrossRef]
- Barba-Spaeth, G.; Longman, R.S.; Albert, M.L.; Rice, C.M. Live attenuated yellow fever 17D infects human DCs and allows for presentation of endogenous and recombinant T cell epitopes. J. Exp. Med. 2005, 202, 1179–1184. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Garratt, R.C.; Caufour, P.S. Surface expression of an immunodominant malaria protein B cell epitope by yellow fever virus. J. Mol. Biol. 2002, 315, 873–885. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Garratt, R.C.; Marchevsky, R.S. Attenuation of Recombinant Yellow Fever 17D Viruses Expressing Foreign Protein Epitopes at the Surface. J. Virol. 2005, 79, 8602–8613. [Google Scholar]
- Tao, D.; Barba-Spaeth, G.; Rai, U.; Nussenzweig, V.; Rice, C.M.; Nussenzweig, R.S. Yellow fever 17D as a vaccine vector for microbial CTL epitopes: protection in a rodent malaria model. J. Exp. Med. 2005, 201, 201–209. [Google Scholar]
- Stoyanov, C.T.; Boscardin, S.B.; Deroubaix, S. Immunogenicity and protective efficacy of a recombinant yellow fever vaccine against the murine malarial parasite Plasmodium yoelii. Vaccine 2010. In Press, Corrected Proof. [Google Scholar]
- Editorial. YF17D vector for Lassa fever vaccine. International Innovation 2010, 77–79.
- Bonaldo, M.; Mello, S.; Trindade, G. Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virology Journal 2007, 4, 115. [Google Scholar]
- Bonaldo, M.C.; Martins, M.A.; Rudersdorf, R. Recombinant Yellow Fever Vaccine Virus 17D Expressing Simian Immunodeficiency Virus SIVmac239 Gag Induces SIV-Specific CD8+ T-Cell Responses in Rhesus Macaques. J. Virol. 2010, 84, 3699–3706. [Google Scholar]
- Stoyanov, C.T.; Boscardin, S.B.; Deroubaix, S. Immunogenicity and protective efficacy of a recombinant yellow fever vaccine against the murine malarial parasite Plasmodium yoelii. Vaccine 2010, 28, 4644–4652. [Google Scholar] [CrossRef]
- Barban, V.; Girerd, Y.; Aguirre, M. High stability of yellow fever 17D-204 vaccine: A 12-year restrospective analysis of large-scale production. Vaccine 2007, 25, 2941–2950. [Google Scholar]
- Monath, T.P.; Myers, G.A.; Beck, R.A. Safety testing for neurovirulence of novel live, attenuated flavivirus vaccines: Infant mice provide an accurate surrogate for the test in monkeys. Biologicals 2005, 33, 131–144. [Google Scholar] [CrossRef]
- Monath, T.P.; Levenbook, I.; Soike, K. Chimeric Yellow Fever Virus 17D-Japanese Encephalitis Virus Vaccine: Dose-Response Effectiveness and Extended Safety Testing in Rhesus Monkeys. J. Virol. 2000, 74, 1742–1751. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; Mitchell, S.W.; Sasso, D.R.; Lange, J.V.; Ramsey, R.; McCormick, J.B. Physiological and immunologic disturbances associated with shock in a primate model of Lassa fever. J. Infect. Dis. 1987, 155, 465–467. [Google Scholar] [CrossRef]
- Peters, C.J. Arenaviruses; Richman, D.D., Whitley, R.J., Eds.; Churchill Livingstone: New York, 1997; pp. 779–799. [Google Scholar]
- Walker, D.H.; Wulff, H.; Lange, J.V.; Murphy, F.A. Comparative pathology of Lassa virus infection in monkeys, guinea-pigs, and Mastomys natalensis. Bull. World Health Organ. 1975, 52, 523–534. [Google Scholar]
- Hensley, L.E.; Smith, M.A.; Geisbert, J.B. Pathogenesis of lassa fever in cynomolgus macaques. Virol. J. 2011, 8, 205. [Google Scholar]
- Carrion, R.J.; Patterson, J.L. Vaccines against viral hemorrhagic fevers: non-human primate models. Hum. Vaccin. 2011, 7, 667–673. [Google Scholar] [CrossRef]
- Mansfield, K. Marmoset model commonly used in biomedical research. Comp. Medicine 2003, 53, 383–392. [Google Scholar]
- Gowen, B.B.; Holbrook, M.R. Animal models of highly pathogenic RNA viral infections: Hemorrhagic fever viruses. Antivir. Res. 2008, 78, 79–90. [Google Scholar]
- Omatsu, T.; Moi, M.L.; Hirayama, T.; Takasaki, T.; Nakamura, S.; Tajima, S.; Ito, M.; Yoshida, T.; Saito, A.; Katakai, Y.; Akari, H.; Kurane, I. Common marmoset (Callithrix jacchus) as a primate model of dengue virus infection: development of high levels of viraemia and demonstration of protective immunity. J. Gen. Virol. 2011, 92, 2272–2280. [Google Scholar] [CrossRef]
- Firsova, I.; Shatokhina, I.V.; Borisevich, I.V.; Evseev, A.A.; Maksimov, V.A.; Pantiukhov, V.B.; Khmelev, A.L. Use of guinea pigs for assessing the efficacy of vaccines against Lassa fever. Vopr Virusol. 2003, 48, 43–45. [Google Scholar]
- Flatz, L.; Rieger, T.; Merkler, D.; Bergthaler, A.; Regen, T.; Schedensack, M.; Bestmann, L.; Verschoor, A.; Kreutzfeldt, M.; Brück, W. T cell-dependence of Lassa fever pathogenesis. PLoS Pathogen. 2010, 6, e1000836. [Google Scholar]
- Yun, N.E.; Poussard, A.L.; Seregin, A.V.; Walker, A.G.; Smith, J.K.; Aronson, J.F.; Smith, J.N.; Soong, L.; Paessler, S. Functional Interferon System Is Required for Clearance of Lassa Virus. J. Virol. 2012, 86, 3389–3392. [Google Scholar]
- Schildknecht, A.; Welti, S.; Geuking, M.B.; Hangartner, L.; van den Broek, M. Absence of CTL Responses to Early Viral Antigens Facilitates Viral Persistence. J. Immunol. 2008, 180, 3113–3121. [Google Scholar]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa virus nucleoprotein mutants generated by reverse genetics induce robust type I IFN response in human dendritic cells and macrophages. J. Virol. 2011, 85, 12093–12097. [Google Scholar] [CrossRef]
- Marzi, A.; Feldmann, H.; Geisbert, T.W.; Falzarano, D. Vesicular Stomatitis Virus-Based Vaccines for Prophylaxis and Treatment of Filovirus Infections. J. Bioterror. Biodef. 2011. [Google Scholar]
- Appaiahgari, M.B.S.V. Clinical development of IMOJEV ® - a recombinant Japanese encephalitis chimeric vaccine (JE-CV). Exp. Opin. Biol. Ther. 2012. [Google Scholar]
- Guy, B.; Barrere, B.; Malinowski, C.; Saville, M.; Teyssou, R.; Lang, J. From research to phase III: Preclinical, industrial and clinical development of the Sanofi Pasteur tetravalent dengue vaccine. Vaccine 2011, 29, 7229–7241. [Google Scholar]
- Leroy, E.M.; Gonzale, J.P.; Baize, S. Ebola and Marburg haemorrhagic fever viruses: major scientific advances, but a relatively minor public health threat for Africa. Clin. Microbiol. Infect. 2011, 17, 964–976. [Google Scholar]
- McKee, K.T.J.; Enria, D.A.; Barerra-Oro, J.G. Vaccines for Biodefense and Energing and Neglected Diseases. In Junin (Argentine Hemorrhagic Fever); Barett, A.D.T., Stanberry, L.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 537–550. [Google Scholar]
- Anonymous. Arboviruses and Related Zoonotic Viruses. 2007. Available online: www.cdc.gov/biosafety/publications/bmbl5/BMBL5_sect_VIII_f.pdf.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Lukashevich, I.S. Advanced Vaccine Candidates for Lassa Fever. Viruses 2012, 4, 2514-2557. https://doi.org/10.3390/v4112514
AMA Style
Lukashevich IS. Advanced Vaccine Candidates for Lassa Fever. Viruses. 2012; 4(11):2514-2557. https://doi.org/10.3390/v4112514
Chicago/Turabian StyleLukashevich, Igor S. 2012. "Advanced Vaccine Candidates for Lassa Fever" Viruses 4, no. 11: 2514-2557. https://doi.org/10.3390/v4112514